





研究背景
芳基硼酸及其衍生物是广泛可得且具有高度实用性的芳基化试剂。在金属催化剂存在或不存在的情况下,芳基硼酸类化合物已被广泛应用于在原位形成碳-碳和碳-杂原子键(如图1所示,左图)。尽管在概念上和实际应用中,这些方法已经被证明是有价值的,但追求一种能够在芳基硼酸的原位/邻位上同时实现双官能化的替代方法,将为有机合成领域带来更多的发展机遇(如图1所示,右图)。
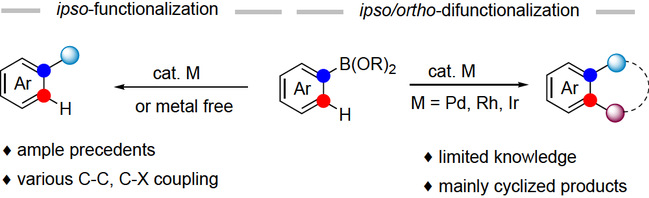
图1 芳基硼酸的转化
在芳基硼酸的原位/邻位双官能化中,反应的关键在于如何将金属催化剂引入到芳基硼酸的邻位。Miura课题组采用merry-go-round策略,用张力烯烃实现芳基硼酸的多次烷基化反应。该反应对烯烃底物的限制条件非常严格,因此这种方法在合成应用上受到了限制。
随后,炔烃作为偶联试剂被用于芳基硼酸的原位/邻位双官能团化反应(见图2a)。通常情况下,通过转金属和炔烃插入生成的烯基金属物种I会发生C–H活化反应形成5元环金属中间体II,或者进行1,4-金属迁移至芳基硼酸的邻位形成中间体III。这些中间体分别被另一个炔烃或分子内亲电位点捕获,得到环状的双官能团化产物(见图2a)。Hayashi课题组首次报道了铑催化芳基硼酸与两分子对称内炔的双官能团化反应,合成一系列萘环衍生物。自此以后,化学家一直在努力拓展芳基硼酸的环状原位/邻位双官能团化反应(见图2a)。近年,周强辉、张扬会和董广斌课题组分别报道了钯/降片烯(NBE)催化的苯硼酸参与的Catellani反应,获得了各种非环化的原位、邻位双官能团化产物(图2b)。然而,由于Catellani反应β–C消除对于位阻的需要,这些转化仅限于邻位取代的芳基硼酸,并需要使用大量的降冰片烯。
本研究中,作者受最近通过加成和消除反应实现丙二腈衍生物的C–CN键活化的启示,设想是否可以设计一种基于1,4-铑迁移和官能团迁移的串联过程的催化方案,从而可能完成芳基硼酸的原位/邻位的烯基氰基化反应,构建官能团化的邻烯基芳基腈(图2c)。
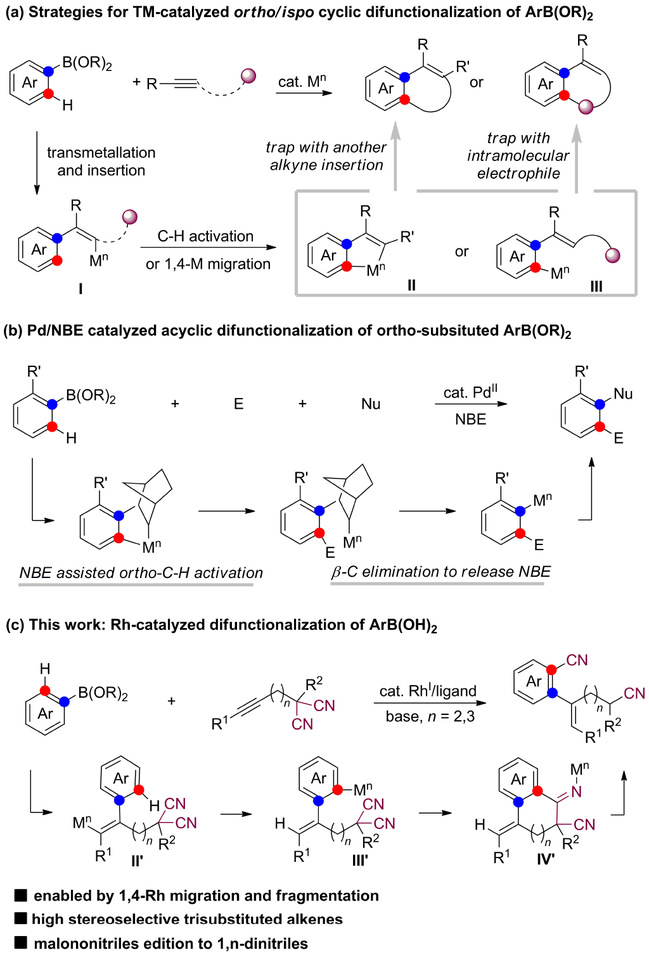
图2 过渡金属催化的芳基硼酸的原位/邻位双官能化
在研究初期存在以下挑战性难题,使得这种策略的实施存在不确定性:(1)迁移插入炔烃反应的区域选择性是否能够按设计发生;(2)芳基铑物种直接与丙二腈位点反应而不是插入C–C三键;(3)化学选择性问题,中间体IV'的氮中心可能会发生竞争性质子化反应,形成环状产物,而不是裂解来实现氰转移。
本研究中,华南理工大学制浆造纸工程国家重点实验室黄良斌教授课题组利用1,4-金属迁移和氰基转移机理为芳基硼酸的非环化双官能团化提供一种新的模式。该方案具有良好的区域选择性和化学选择性、温和的反应条件和广泛的底物适用范围等特点。
图文解读
作者通过对反应条件的筛选,发现当使用10 mol%的BINAP作为配体时,目标产物3a的收率为49%。配体的性质对反应活性有着不可忽略的影响。TFP可以显著提高收率至90%,而DPPB直接降低了反应活性。当使用非极性溶剂,如1,4-dioxane和toluene时,可以获得优异的3a收率。极性溶剂DMF未能得到期望的产物。同样,碱也反应产生了显著的影响。碳酸钾是这个反应的优良碱,然而使用KF作为碱时没有检测到目标产物。当反应温度降至60°C时,产物收率稍有提高。因此,最佳反应条件A为芳基硼酸1a(0.15 mmol),炔基二腈2a(0.1 mmol),RhCl(PPh3)3 (5 mol%, 0.05 mmol),TFP(10 mol%, 0.01 mmol),K3PO4 (0.2 mmol),toluene(1 mL),氮气氛围下60°C搅拌6小时。
表1 条件筛选


a) Reaction conditions: 1a (0.15 mmol, 1.5 equiv.), 2a (0.10 mmol, 1.0 equiv.), base (0.20 mmol, 2 equiv.), RhCl(PPh3)3 (5.0 mol%), ligand (10 mol%), solvent (0.10 M, 1 mL) under 100 °C, b) Corrected GC yields were determined by using dodecane as an internal standard, c) 60 °C instead of 100 °C. Isolated yield in the parentheses. TFP is tri(furan-2-yl)phosphane, DPPB is 1,4-bis(diphenylphosphanyl)butane.
在最优的反应条件下,作者首先对芳基硼酸底物的适用范围进行了探索。如图3所示,芳基硼酸苯环上的取代基不管是富电子基团、贫电子基团和电中性基团都能顺利地反应,以中等偏上的产率得到目标产物。多种官能团在该反应体系中都能兼容,包括:腈、醛、酮、酯。对于带有邻位取代基的芳基硼酸,氰转移选择性地发生在立体位阻较小的位置,这是由于1,4-铑迁移发生在立体位阻较小的位置。此外除了单取代,二取代芳基硼酸底物在该体系中也能很好地兼容。作者发现含有喹啉、苯并噻吩和二苯并呋喃的杂环芳基硼酸的烯基氰基化反应进行顺利,产物收率中等到良好。螺环芳基硼酸在反应中兼容,该底物可能在有机发光二极管中有重要应用。
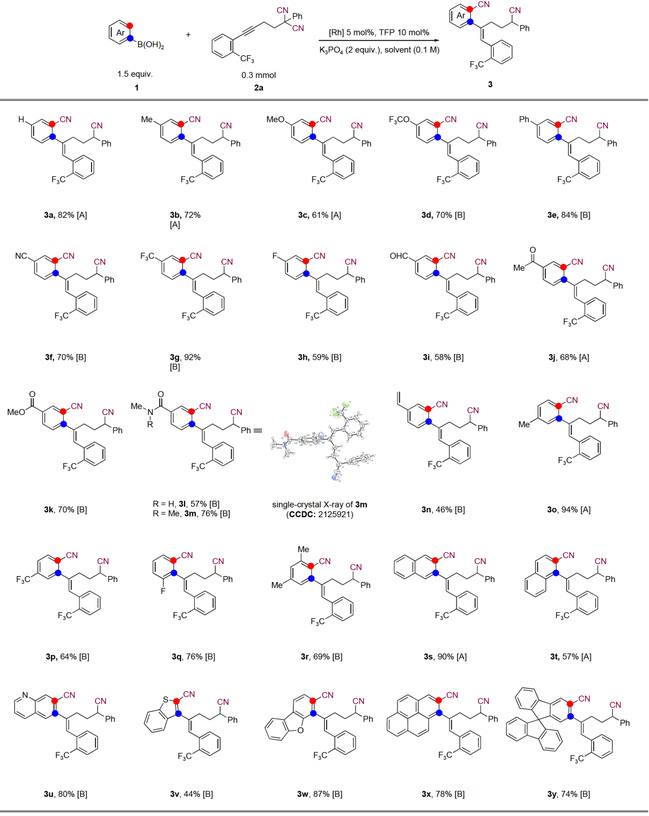
Reaction conditions A: RhCl(PPh3)3 (5 mol%), toluene (0.1 M) at 60 °C in N2 for 6 h, Reaction conditions B: [RhCl(cod)]2 (2.5 mol%), dioxane (0.1 M) at 100 °C in N2 for 20 h.
图3 芳基硼酸的底物拓展
随后,作者对炔基二腈底物类型进行了拓展(图4)。由于需要空间位阻来区分炔的两侧,因此炔基二腈的芳基上的有空间位阻的取代基对于区域选择素的迁移插入至关重要。各种邻芳基取代的炔基二腈具有良好的耐受性,并以中等到良好的产率转化为相应的产物。然而当芳基对位上有一个氰基时,同样以中等收率得到了目标产物,这可能是由于氰基的电子效应控制了炔插入的区域选择性。缺电子的杂环芳基取代的炔烃,如吡啶和喹啉都具有良好的耐受性。作者同时也对炔基二腈α位取代基的普适性进行考察,带有取代基的芳基和烯基都与反应相容。
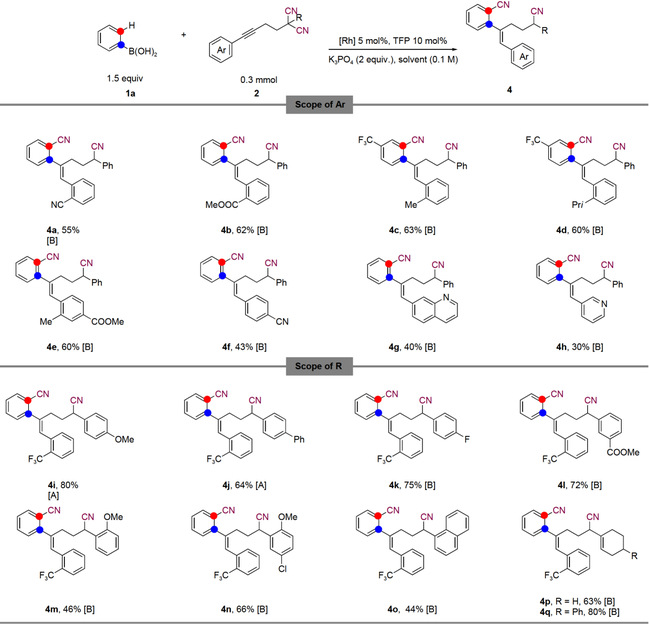
Reaction conditions A: RhCl(PPh3)3 (5 mol%), toluene (0.1 M) at 60 °C in N2 for 6 h, Reaction conditions B: [RhCl(cod)]2 (2.5 mol%), dioxane (0.1 M) at 100 °C in N2 for 20 h.
图4 炔基二腈底物拓展
接下来,作者探索了反应活性较低的α位苄基取代基的影响(图5)。在100℃下得到了混合物,氰基转移产物4r和环庚酮产物5a的比例为3:1。作者通过温度控制来调整选择性,在40℃时选择性得到了5a;当温度逐渐升高至140℃时,5a完全被抑制,4r以78%的收率得到。苄基和芳基取代底物之间的不同反应性可以通过炔基二腈α位的酸性来解释,在某种程度上酸性增强可能会促使β–C消除。
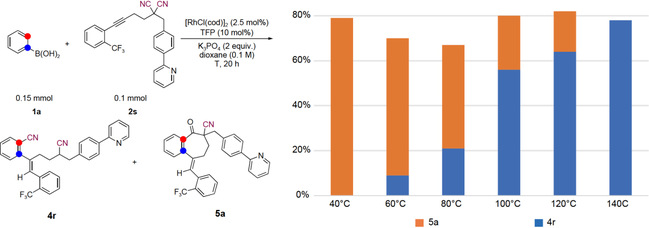
图5 温度调控苄基取代炔基二腈的氰基转移
在找到最佳反应条件后,作者探索了各种苄基取代的炔基二腈的适用范围(图6)。各种苄基取代基的炔基二腈均为合适的底物。作者同时也对氰基转移过程的碳链长度进行了考察,增长一个碳链的底物结果收率较低,因为反应经历一个更不稳定的8元环中间体。具有更长碳链的底物在该反应中不兼容。短链底物没有反应,因为在这种情况下炔基两端的位阻增加,迁移插入的过程很难发生。作者还探究了40℃合成环庚酮的反应范围。多种烷基取代的炔基二腈是合适的底物。作者通过尝试几种手性配体来探索5b的不对称合成,并得到了一个有希望的结果,70%的收率和55%ee。
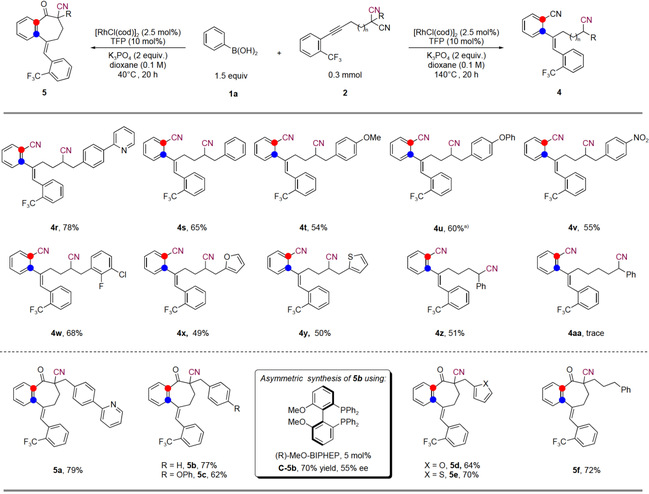
Unless otherwise noted, all reactions were run with 1a (0.45 mmol), 2 (0.3 mmol), [RhCl(cod)]2 (2.5 mol%), TFP (10 mol%), K3PO4 (2 equiv.) and dioxane (0.1 M) at 140 or 40 °C in N2 for 20 h, a) 160 °C instead of 140 °C.
图6 苄基取代炔基二腈的底物拓展
为了深入了解这些转化的机理,作者进行了一些控制实验(图7)。将丙二腈的一个氰基换成酯基后,作者只观察到微量的氰转移产物6,并且得到了复杂的混合物。这表明丙二腈基团对反应至关重要(图7a)。作者调查了带有单取代基的丙二腈2ac是否可以参与反应,结果发现反应无法发生。这表明具有季碳中心的底物对反应至关重要(图7b)。此外,还进行了氘标记实验(图7c),d-1a的邻位氘到转移到产物的烯基位置,证明了反应发生了1,4-铑(I)迁移。作者探索了非芳基取代的炔基二腈是否可以作为氰基转移的底物。烷基取代的炔基二腈迁移插入的区域选择性显著降低。TMS取代的炔基二腈2af区域选择性较好,仅获得具有1,4-Rh迁移的氰基转移产物10a但收率较低(图7d)。
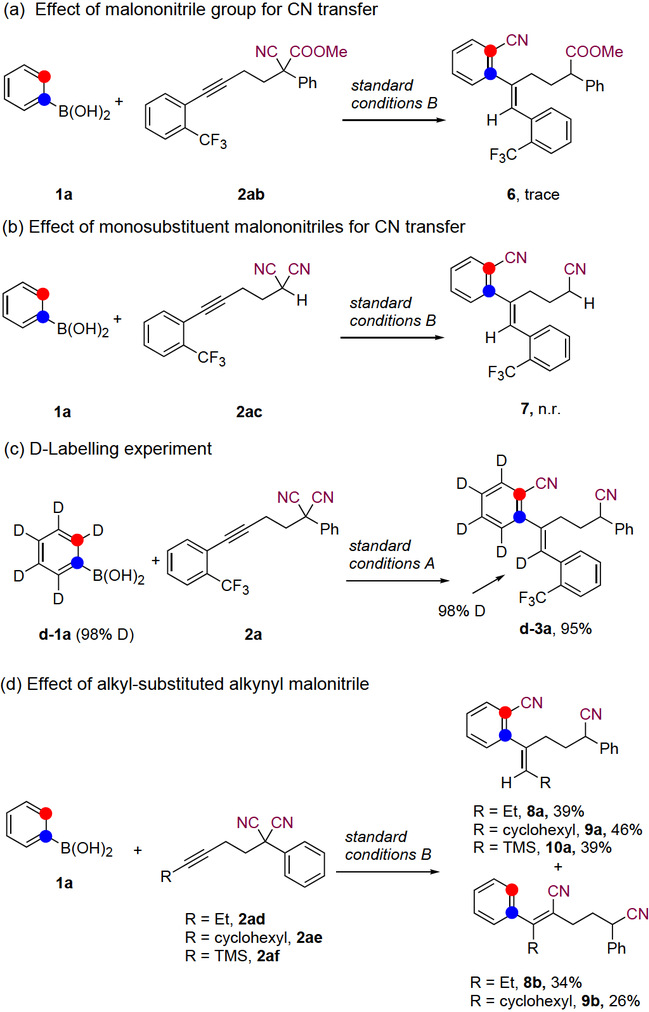
图7 控制实验
将上述模板反应进行放大,在较低催化剂负载下,也能获得86%的产物,体现了该反应策略的实用性(图8,顶部)。所得产物可以很方便地转化为其他有价值的分子(图8)。在苯乙烯作为HCN受体的存在下,发生了脱氢氰化反应,然后异构化产生了1,3-二烯产物11(图8a)。苄基腈的C–CN活化发生了Heck类型的反应(图8b)。利用辛-4-炔作为HCN受体,3a通过双C–CN键活化和环化转化为萘13(图8c)。作者利用苄基腈α位C–H键的酸性与丙烯基溴和TMSCF2Br偶联得到相应的产物(图8d-e)。此外,芳基和苄基腈都可以水合成酰胺16(图8f)。
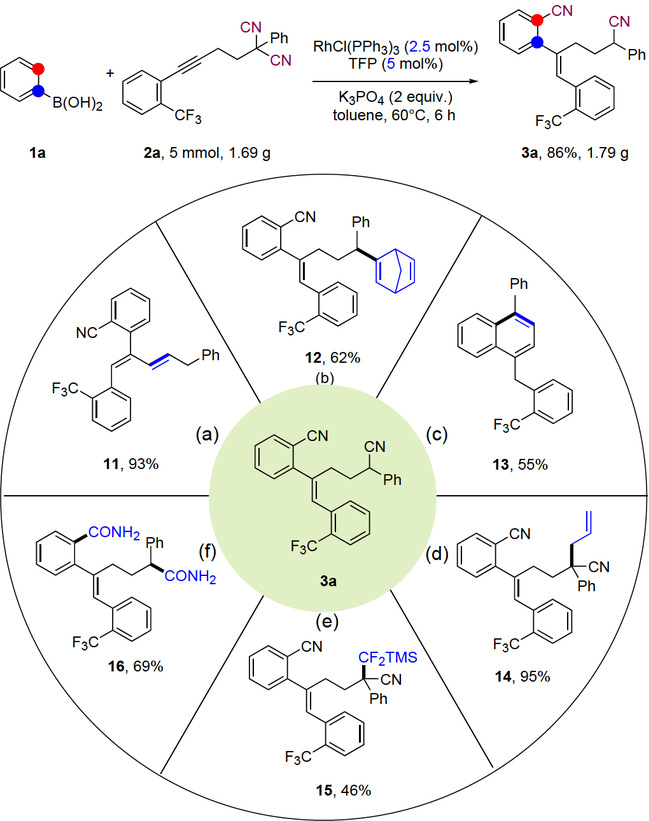
Reaction conditions: (a) Pd2(dba)3 (2.5 mol%), CyJohnphos (10 mol%), AlMe2Cl (0.2 equiv.), styrene (5.0 equiv.), toluene, 100 °C, N2, 16 h; (b) Ni(cod)2 (5 mol%), DPEphos (5 mol%), AlMe2Cl (0.2 equiv.), 2,5-Norbornadiene (1.1 equiv.), toluene, rt., N2, 16 h; (c) Ni(cod)2 (5 mol%), DPEphos (5 mol%), AlMe2Cl (0.2 equiv.), oct-4-yne (1.1 equiv.), toluene, 80 °C, N2, 16 h; (d) LDA (1.2 equiv.), Allyl bromide (1.2 equiv.), THF, 0 °C, N2, 10 h; (e) nBu-Li (1.2 equiv.), TMSCF2Br (3.0 equiv.), toluene, rt., 5h; (f) 30% H2O2 (0.05 M), K2CO3 (2 equiv.), DMSO, rt., 16 h.
图8 克级反应以及合成应用
基于上述的控制实验,作者提出了一种可行的反应机理(图9)。首先,通过芳基硼酸与铑催化剂I的转金属作用生成芳基铑物种II。Ar–Rh物种迁移插入到炔烃2中形成烯基铑物种III,然后发生1,4-迁移。由此得到的芳基铑中间体IV与一个氰基配位,然后插入到氰基中生成中间体V。最后,V的β–C消除和质子化获得目标产物3。中间体V竞争性质子化生成亚胺,亚胺进一步水解为环庚酮衍生物5。
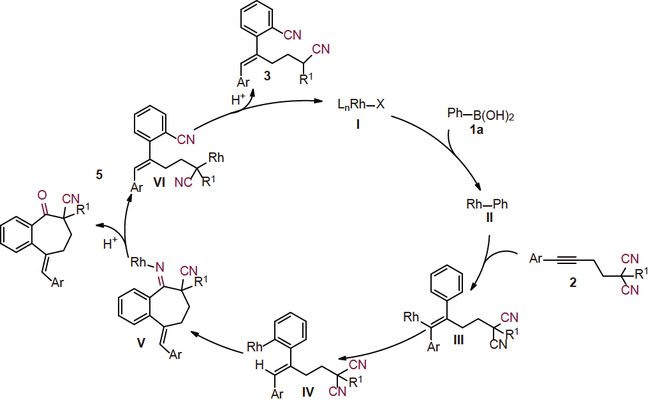
图9 推测的反应机理
论文信息
相关研究成果以“Redox-Neutral ipso/ortho Alkenylcyanation of (hetero)Arylboronic Acid Enabled by 1,4-Rhodium Migration and Fragmentation”为题,在线发表于《Science China Chemistry》。文章第一作者为华南理工大学硕士研究生郭晨霞,通讯作者为华南理工大学制浆造纸工程国家重点实验室黄良斌教授。
原文链接:https://link.springer.com/article/10.1007/s11426-023-1645-x