朱伟教授团队在Nature子刊STTT发表论文:巨噬细胞代谢重编程, 攻克糖尿病慢性创面修复难题
糖尿病是全球重大公共卫生问题,糖尿病足溃疡(DFU)作为其最严重的慢性并发症,是患者截肢、死亡的首要诱因。持续高血糖引发血管功能障碍、周围神经病变及免疫失衡,导致创面再上皮化受阻、感染风险高、愈合延迟且复发率高,给患者带来巨大痛苦与医疗负担。
巨噬细胞是先天免疫核心调控者和创面愈合关键枢纽,正常愈合中会动态转换表型:炎症早期以M1型为主,负责清除病原体、启动炎症;消退期转为M2修复型,主导组织再生、胶原沉积及血管新生。这一转换受代谢调控,M1型依赖糖酵解供能,M2型则高度依赖线粒体氧化磷酸化(OXPHOS)。但糖尿病高糖微环境下,胰岛素抵抗破坏巨噬细胞葡萄糖代谢稳态,阻断M1向M2转换,使其长期处于促炎M1状态,导致创面陷入慢性炎症恶性循环。现有治疗手段多聚焦于调控巨噬细胞免疫表型,无法从根源纠正代谢紊乱,难以实现长效修复,临床疗效有限。
钒作为人体必需微量元素,具有类胰岛素活性,可精准调控葡萄糖代谢并兼具免疫调节功能,是破解该代谢紊乱的理想靶点。但此前其在糖尿病创面修复中的应用未被系统挖掘,且钒离子的控释难题及剂量依赖性全身毒性,严重制约了临床转化。
基于以上核心临床与科学痛点,华南理工大学生物科学与工程学院朱伟教授联合陆军军医大学西南医院全军烧伤研究所罗高兴教授和于云龙研究员创新性地将钒元素与介孔生物活性玻璃结合,开发出兼具代谢调控与免疫调节功能的V-MBG纳米材料,并配套构建了葡萄糖响应性水凝胶递送平台,实现了钒离子在创面微环境的“按需释放”与精准治疗。
相关研究成果以Macrophage metabolic reprogramming by vanadium released from glucose-responsive bio-gel accelerates diabetic wound repair为题,于2026年4月23日在线发表于国际顶级医学期刊Signal Transduction and Targeted Therapy(IF=52.7)。
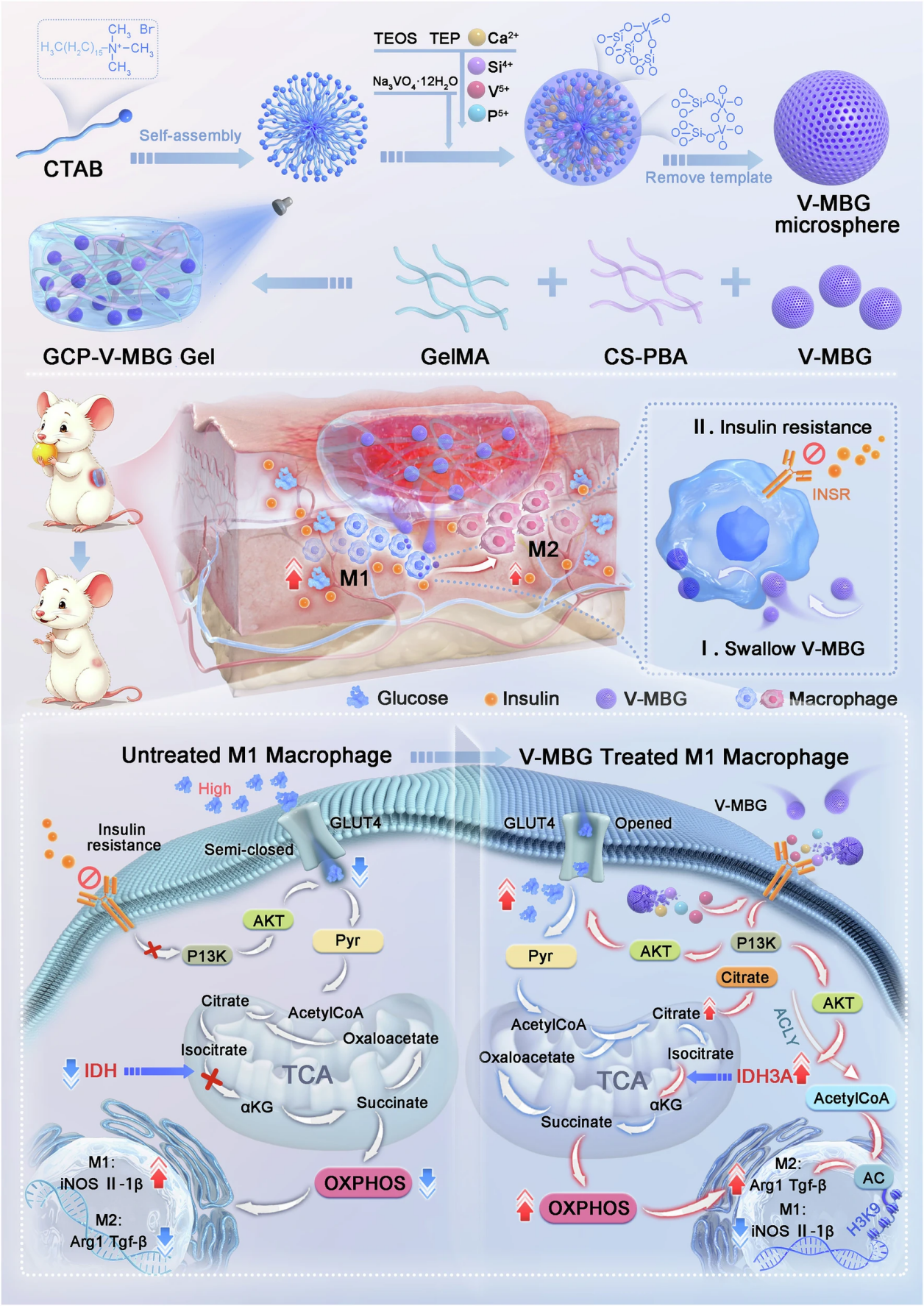
图1. V-MBG纳米微球合成、巨噬细胞代谢调控机制及糖尿病创面治疗应用
该示意图完整呈现了研究的核心设计逻辑,从V-MBG纳米微球的溶胶-凝胶合成路线,到其被巨噬细胞吞噬后通过激活INSR-PI3K信号轴实现代谢重编程、驱动M2极化的分子机制,再到葡萄糖响应性水凝胶在糖尿病创面中的治疗应用。
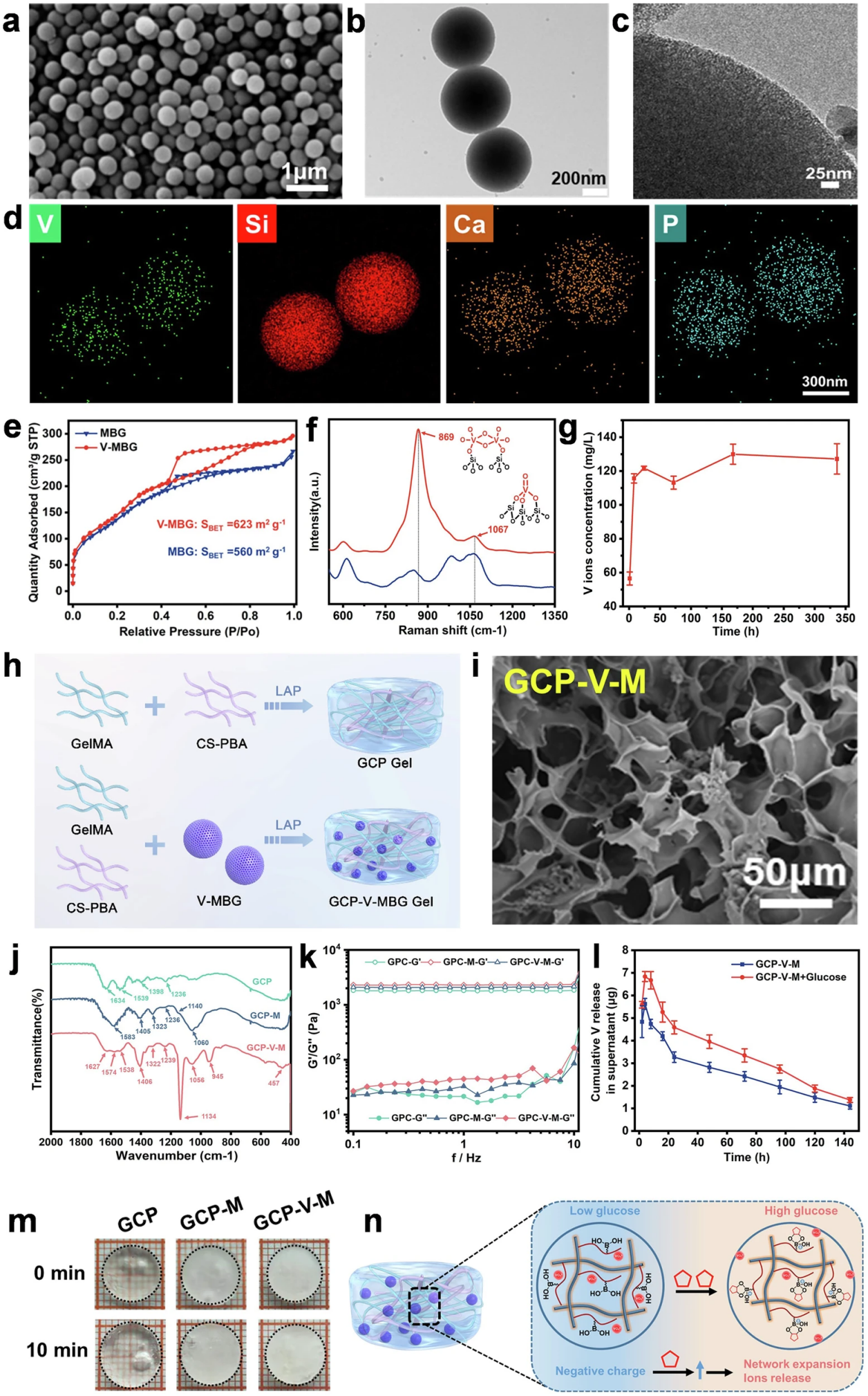
图2. V-MBG纳米微球与GCP-V-MBG葡萄糖响应性水凝胶的理化性质表征
研究团队通过改良溶胶-凝胶法,以十六烷基三甲基溴化铵(CTAB)为模板,成功合成了基于CaO-SiO2-P2O5体系的V-MBG纳米微球。该微球为粒径均一的球形纳米颗粒(约500±20.5 nm),具有规整有序的介孔结构,钒、硅、钙、磷元素实现均匀分布,比表面积高达623 m2/g,钒离子可在体外实现持续、平稳缓释。
为实现钒离子的靶向可控递送,团队将葡萄糖敏感型苯硼酸(PBA)接枝到羧甲基壳聚糖制备CS-PBA,与甲基丙烯酸明胶(GelMA)共同构建光交联GCP水凝胶网络,包载V-MBG后获得GCP-V-MBG(GCP-V-M)水凝胶。表征结果显示,V-MBG的掺入显著提升了水凝胶的机械强度,且未改变其三维多孔形貌;在模拟糖尿病创面的高糖环境中,该水凝胶可通过葡萄糖触发的基质膨胀效应,实现钒离子浓度依赖性的“按需释放”,既保证局部治疗浓度,又最大限度降低了全身暴露的毒性风险,解决了钒基制剂临床转化的核心瓶颈。
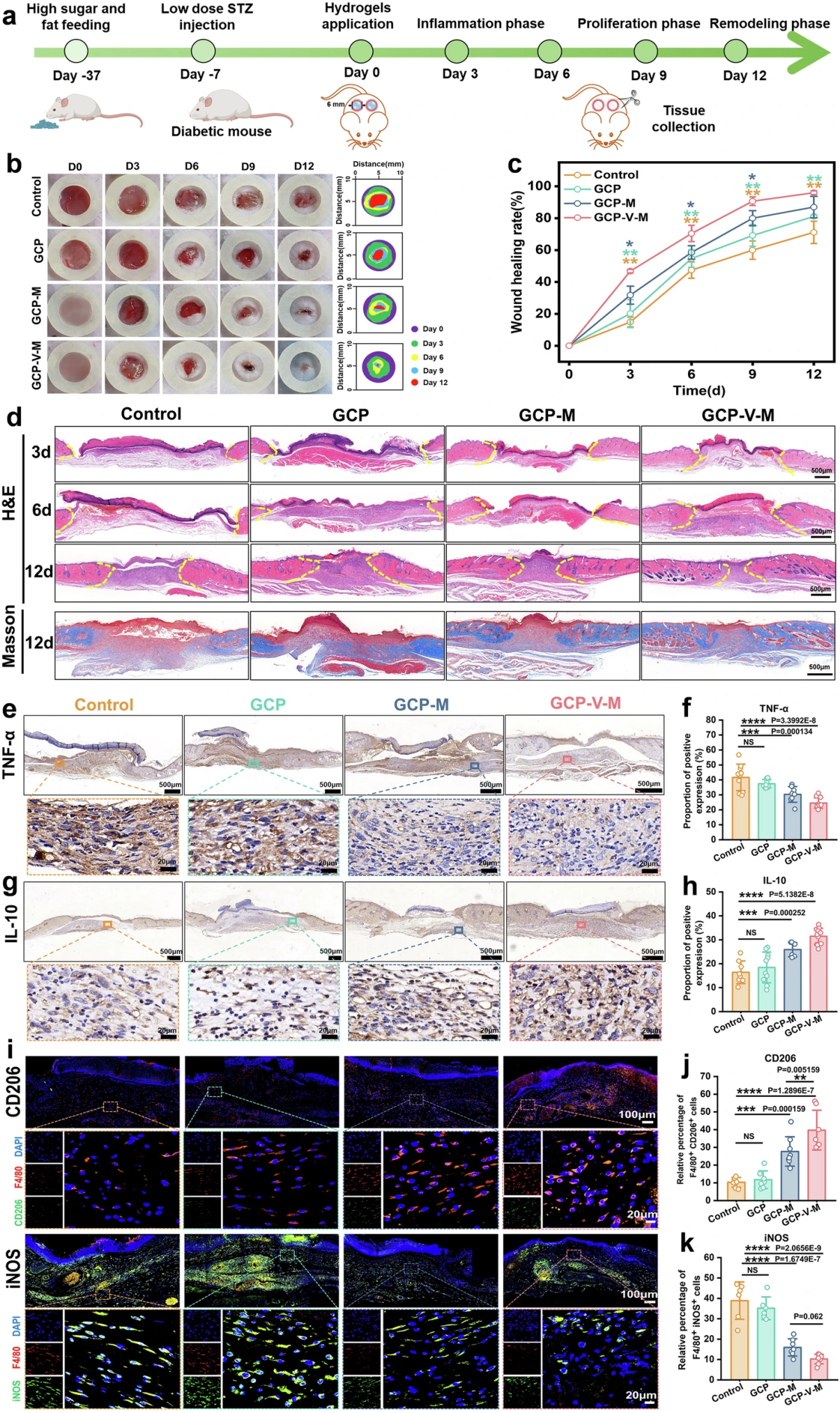
图3. GCP-V-MBG水凝胶促进糖尿病小鼠创面愈合的体内疗效与机制验证
研究团队通过高脂高糖饮食联合低剂量链脲佐菌素(STZ)注射,成功构建稳定的C57小鼠糖尿病模型,非靶向代谢组学证实小鼠创面组织存在显著代谢紊乱,完美模拟了临床糖尿病创面的病理特征。团队在小鼠背部构建6 mm全层皮肤缺损模型,分为空白对照组、GCP空白水凝胶组、GCP-MBG水凝胶组与GCP-V-M水凝胶组开展治疗实验。
结果显示,GCP-V-M治疗组在各时间点均显著加速创面闭合,治疗12天时创面愈合率高达96.00±1.18%,实现近完全愈合,疗效显著优于所有对照组。组织学染色验证了其优异的修复效果:H&E染色显示,GCP-V-M组治疗3天和6天时表皮与肉芽组织层更厚,提示早期炎症响应与再生进程被有效激活;治疗12天时表皮与肉芽组织层显著变薄,表明创面更快进入成熟重塑阶段。Masson三色染色证实,GCP-V-M治疗组创面胶原沉积量显著提升,胶原排列更规整,为创面愈合提供了良好的结构支撑。
免疫组化与免疫荧光染色进一步解析了核心治疗机制:GCP-V-M治疗可显著下调创面促炎因子TNF-α、IL-6的表达,大幅上调抗炎因子IL-10的表达,有效抑制创面过度慢性炎症;CD31染色结果证实,该水凝胶还显著促进了创面新生血管形成,为组织再生提供充足血供。针对巨噬细胞的检测结果显示,治疗4天时,GCP-V-M组创面CD206阳性M2型巨噬细胞比例显著升高,iNOS阳性M1型巨噬细胞比例显著降低,证实该水凝胶可在体内高效驱动巨噬细胞向修复型M2表型极化,从根源重塑了创面的抗炎修复微环境。
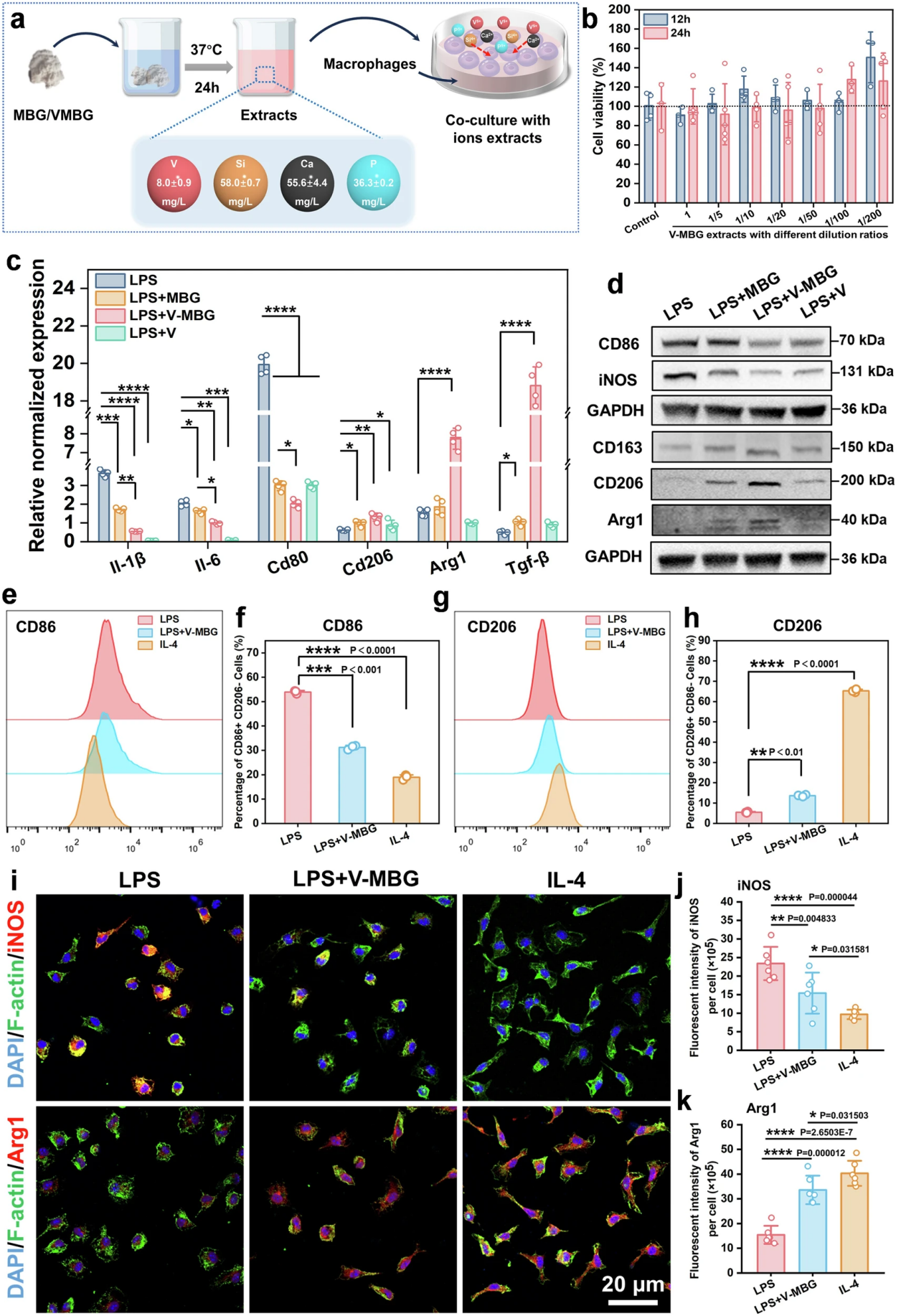
图4. V-MBG 体外调控巨噬细胞向M2表型极化的直接验证
为明确V-MBG调控巨噬细胞极化的直接作用,团队开展了系统的体外细胞实验。制备的V-MBG与MBG浸提液经ICP检测证实,二者硅、钙、磷浓度无显著差异,钒离子为V-MBG特有,排除了其他离子的干扰。CCK-8与活/死染色实验证实,V-MBG浸提液在实验浓度下无明显细胞毒性,特定稀释比下还可轻微提升巨噬细胞活力,具备良好的生物相容性。
在LPS诱导的M1型巨噬细胞模型中,RT-qPCR结果显示,V-MBG处理可显著下调Il-1β、Il-6、Cd80等M1相关基因,同时上调Cd206、Arg1、Tgf-β等M2相关基因;蛋白免疫印迹实验进一步在蛋白水平验证了该结果,V-MBG处理显著降低CD86、iNOS等M1标志物,同时提升CD163、CD206、Arg1等M2标志物的表达。流式细胞术与免疫荧光染色结果证实,V-MBG处理可显著减少CD86+/CD206-的M1型巨噬细胞群,增加CD206+/CD86-的M2型巨噬细胞群,其促M2极化效应与经典M2诱导剂IL-4相当,直接验证了V-MBG驱动巨噬细胞表型转换的核心作用。
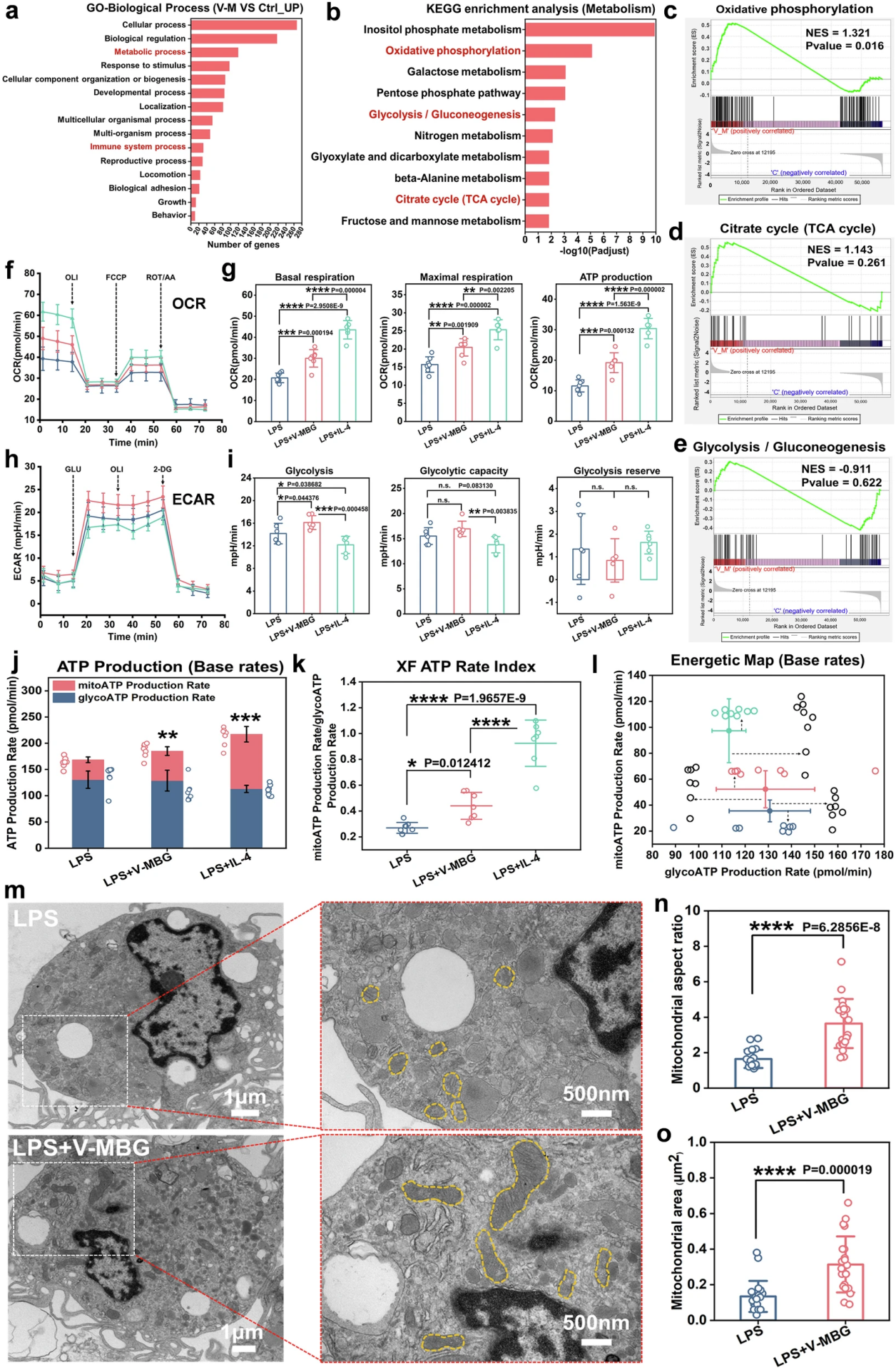
图5. V-MBG重塑巨噬细胞能量代谢,向OXPHOS主导型表型转换
为揭示V-MBG驱动巨噬细胞极化的核心代谢机制,团队开展了转录组学与功能代谢实验。转录组测序结果显示,V-MBG处理组与对照组巨噬细胞的差异表达基因显著富集于“代谢过程”与“免疫系统过程”;KEGG富集分析发现,氧化磷酸化、糖酵解/糖异生、三羧酸(TCA)循环等核心能量代谢通路被显著激活,提示V-MBG通过重塑巨噬细胞能量代谢驱动表型转换。
Seahorse能量代谢检测结果显示,V-MBG处理显著提升了巨噬细胞的氧气消耗速率(OCR),基础呼吸、最大呼吸能力与ATP生成量均大幅升高,线粒体氧化磷酸化水平与IL-4处理组相当;而细胞外酸化率(ECAR)检测显示,V-MBG仅轻微提升糖酵解活性,远不及对线粒体呼吸的促进效应。ATP生成速率分析证实,V-MBG处理组总ATP生成量显著升高,且主要来源于OXPHOS,巨噬细胞完成了向OXPHOS主导的代谢转换。透射电镜观察结果提供了结构层面的证据:V-MBG处理后,巨噬细胞内线粒体体积增大、融合程度提升、膜表面积增加,线粒体纵横比与面积均显著升高,为OXPHOS水平的提升提供了直接的结构基础。
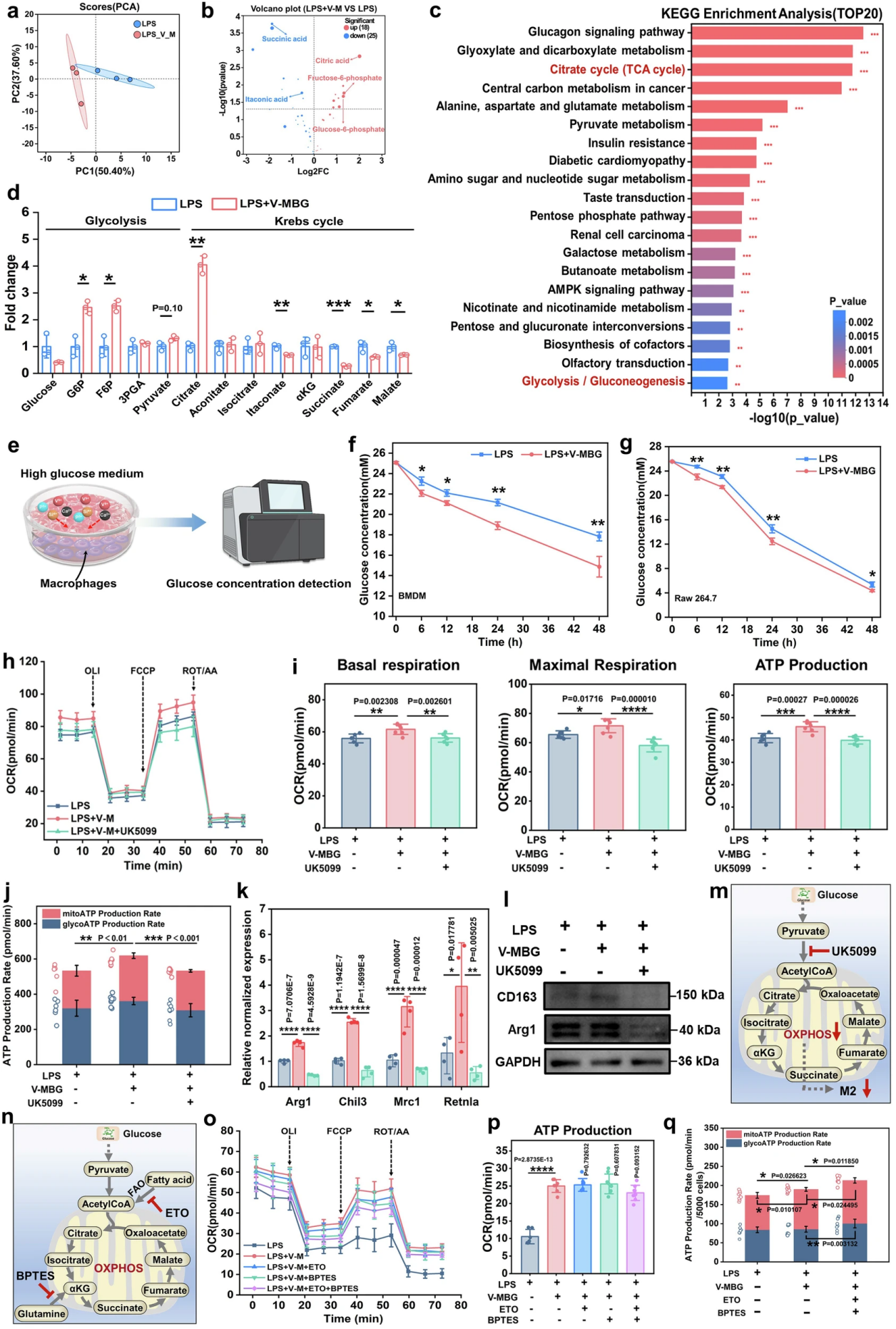
图6. 葡萄糖驱动的线粒体OXPHOS是V-MBG诱导巨噬细胞M2极化的核心必要条件
团队通过代谢组学与功能阻断实验,最终明确葡萄糖代谢是V-MBG实现巨噬细胞代谢重编程的核心能量来源。中心碳代谢组学分析显示,V-MBG处理组与对照组巨噬细胞的代谢谱存在显著差异,共筛选出43个差异代谢物,主要集中于糖酵解与TCA循环通路。代谢物定量结果显示,V-MBG处理后巨噬细胞内糖酵解中间体G6P、F6P与TCA循环关键物质柠檬酸水平显著升高,衣康酸、琥珀酸等促炎代谢物水平降低,同时胞内葡萄糖含量下降,提示葡萄糖摄取与代谢通量显著提升;时间梯度检测也证实,V-MBG处理显著增强了巨噬细胞的葡萄糖摄取与消耗能力。
功能阻断实验进一步验证了其核心必要性:使用UK5099阻断线粒体丙酮酸输入后,V-MBG诱导的巨噬细胞基础呼吸、最大呼吸与ATP生成均显著下降,OXPHOS来源的ATP大幅减少,同时V-MBG诱导的M2相关基因与蛋白表达被显著抑制,证实线粒体丙酮酸摄取是V-MBG实现代谢重编程与M2极化的关键环节。而抑制剂实验证实,抑制脂肪酸氧化(ETO)与谷氨酰胺分解(BPTES)仅对巨噬细胞线粒体呼吸产生轻微影响,不会显著降低OXPHOS来源的ATP,也无法逆转V-MBG诱导的M2极化,最终明确V-MBG主要以葡萄糖驱动的线粒体OXPHOS为核心能量来源,实现巨噬细胞M2极化。
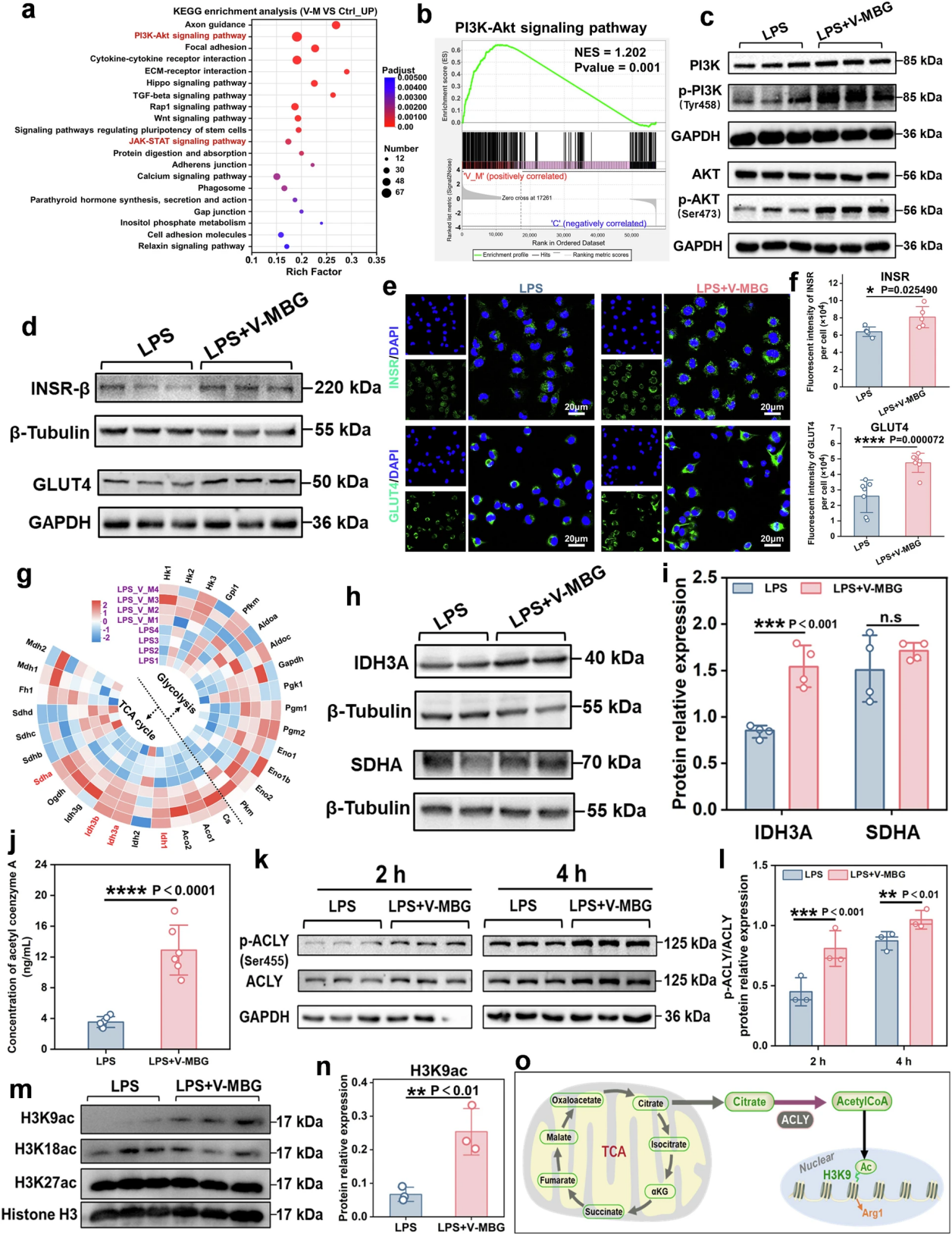
图7. V-MBG驱动巨噬细胞代谢重编程与极化的分子与表观遗传机制
团队通过多组学与分子实验,进一步挖掘了上游分子调控网络。转录组KEGG富集分析与GSEA验证显示,PI3K-AKT通路被显著激活;蛋白免疫印迹实验证实,V-MBG处理后PI3K与AKT的磷酸化水平显著升高,而JAK-STAT通路无明显激活,明确了PI3K-AKT是V-MBG发挥作用的核心信号轴。
进一步研究发现,V-MBG显著上调了胰岛素受体(INSR)的基因与蛋白表达,同时显著提升葡萄糖转运体GLUT4的表达,而GLUT1表达无明显变化;后续抑制剂实验证实,GLUT4是V-MBG诱导巨噬细胞葡萄糖摄取的主要转运体,GLUT1仅发挥代偿作用,明确V-MBG通过激活INSR-PI3K-AKT轴,上调GLUT4表达增强巨噬细胞葡萄糖摄取。同时,该信号轴可通过上调TCA循环关键限速酶IDH3A的表达,恢复被LPS抑制的TCA循环通量,为OXPHOS提供了充足的代谢支撑。
除此之外,团队还发现了关键的表观遗传调控机制:V-MBG处理后巨噬细胞内柠檬酸水平升高,通过激活ATP柠檬酸裂解酶(ACLY)显著提升乙酰辅酶A(Ac-CoA)的含量,进而上调组蛋白H3K9ac的乙酰化水平,通过柠檬酸-Ac-CoA-H3K9ac表观遗传轴,促进M2型相关基因的表达,为巨噬细胞表型转换提供了表观遗传层面的放大效应。
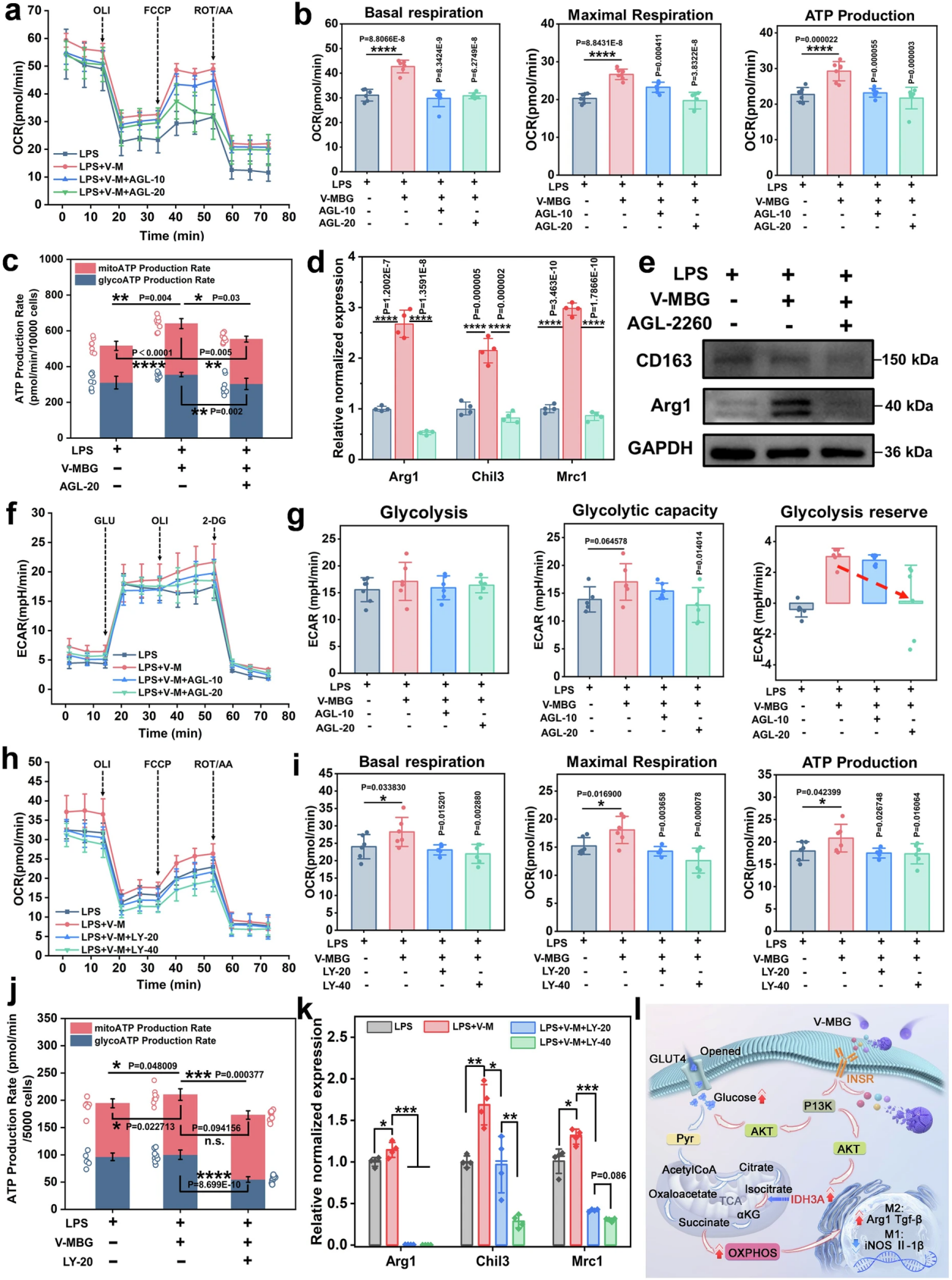
图8. INSR-PI3K信号轴介导葡萄糖驱动的OXPHOS与巨噬细胞M2极化
团队通过系统的抑制剂回补实验,最终完成了核心通路的闭环验证。使用INSR特异性抑制剂AGL-2263处理后,V-MBG诱导的巨噬细胞线粒体基础呼吸、最大呼吸与ATP生成呈剂量依赖性下降,OXPHOS与糖酵解来源的ATP均显著减少,同时V-MBG诱导的M2相关基因与蛋白表达被显著抑制,证实INSR是V-MBG发挥代谢调控与促极化效应的上游核心靶点。使用PI3K抑制剂LY294002处理后,巨噬细胞OCR呈剂量依赖性下降,基础呼吸、最大呼吸与ATP生成显著降低,糖酵解活性也被显著抑制,V-MBG诱导的M2极化效应同样被剂量依赖性逆转。而GLUT4特异性抑制剂Fasentin处理,显著降低了巨噬细胞线粒体呼吸水平,并完全逆转了V-MBG诱导的M2相关基因与蛋白表达上调。
最终,研究完整证实了核心作用机制:V-MBG释放的钒离子,可激活巨噬细胞INSR,进而启动PI3K-AKT信号轴,一方面促进GLUT4介导的葡萄糖摄取,另一方面上调IDH3A表达增强TCA循环通量,最终协同提升葡萄糖驱动的线粒体OXPHOS水平;同时,柠檬酸-Ac-CoA-H3K9ac表观遗传轴进一步放大M2极化效应,最终实现巨噬细胞代谢重编程与修复表型转换,加速糖尿病创面愈合。
研究意义与转化前景:
本研究首次系统揭示了钒元素通过靶向巨噬细胞免疫代谢促进糖尿病伤口修复的作用与完整机制,突破了现有糖尿病创面治疗“重免疫表型调控、轻代谢根源纠正”的长期瓶颈,为慢性创面治疗提供了全新的“免疫-代谢调控”研究范式。
团队开发的葡萄糖响应性GCP-V-M水凝胶,实现了钒离子在创面微环境的按需缓释,在显著提升治疗效果的同时,最大限度降低了钒元素的全身暴露与毒性风险,解决了钒基制剂临床转化的核心痛点。与FDA已批准的干细胞、生长因子类疗法相比,该水凝胶体系为“无细胞、无生长因子”的治疗策略,具有成分明确、稳定性好、可即取即用、成本更低、易于规模化生产等显著优势,为糖尿病足溃疡的临床治疗提供了极具转化潜力的全新方案。
同时,该研究提出的“通过生物材料靶向免疫代谢重编程修复慢性创面”的创新理念,也为其他炎症性疾病、缺血性组织损伤与再生医学领域的研究提供了全新的思路与理论支撑。
本研究得到了国家重点研发计划(2021YFA1101100)、国家自然科学基金(82202459、82472570)、重庆市自然科学基金(CSTB2022NSCQ-MSX0153)、重庆市教育委员会科学技术研究计划重点项目(KJZD-M202512806)、中国博士后科学基金面上项目(2023M734261)、重庆市博士后科研项目特别资助(2022CQBSHTB3043),以及陆军军医大学西南医院“博清拓举”学术人才发展基金(2025BQTJ-1)的资助