学院动态 || 王领副教授课题组开发基于分层信息图神经网络的通用QSAR建模算法
近日,我院王领副教授课题组在美国化学会分子模拟与人工智能权威期刊Journal of Chemical Information and Modeling上发表题为“HiGNN: A Hierarchical Informative Graph Neural Network for Molecular Property Prediction Equipped with Feature-Wise Attention”的研究论文。
药物发现是一个非常漫长和昂贵的过程,开发一种新药平均需要十年以上,花费二十五亿美元。分子性质预测是药物发现领域的核心主题之一,准确预测分子的成药性和生物活性在药物设计和发现中起着关键作用。基于人工智能的QSAR建模方法可以很大程度上减少药物研发对于实验的依赖,快速准确地预测候选化合物的生物活性和ADMET等性质能够帮助化学家在药物研发早期快速筛选潜在成功率更高的化合物,从而节省新药研发周期并降低研究成本。
近些年来,图神经网络(Graph Neural Networks,GNN)在基于图的分子性质预测方面取得了显著进步。目前流行的GNN可被抽象为消息传递神经网络(Message Passing Neural Network, MPNN)范式。MPNN包括消息传递阶段与读出阶段,消息传递阶段学习节点的表征,读出阶段聚合所有的节点表示以得到图的表示。然而,当前基于图的深度学习方法忽略了分子的层次信息和特征通道之间的关系。如图1所示,在该研究中,课题组提出了一个精心设计的分层信息图神经网络框架(Hierarchical Informative Graph Neural Network,HiGNN),其通过同时利用分子图和片段信息来预测分子的性质。首先,借用了神经张量网络的思想在HiGNN框架中设计了一个原子间相互作用的GNN编码块,进行特征提取。其次,在HiGNN框架部署了一个即插即用的特征注意块(Feature-wise Attention Block),以在消息传递阶段之后自适应地重新校准原子特征,它能够选择性地关注重要特征。此外,HiGNN算法经验性地使用了一种基于化学领域知识的分子切割算法BRICS(Breaking of Retrosynthetically Interesting Chemical Substructures),将分子切割成片段,然后将分子图和片段同时输入到GNN编码器以获取全局和分层表示。最后,课题组提出了一种分子-片段相似性机制来研究HiGNN模型在子图级别的可解释性。
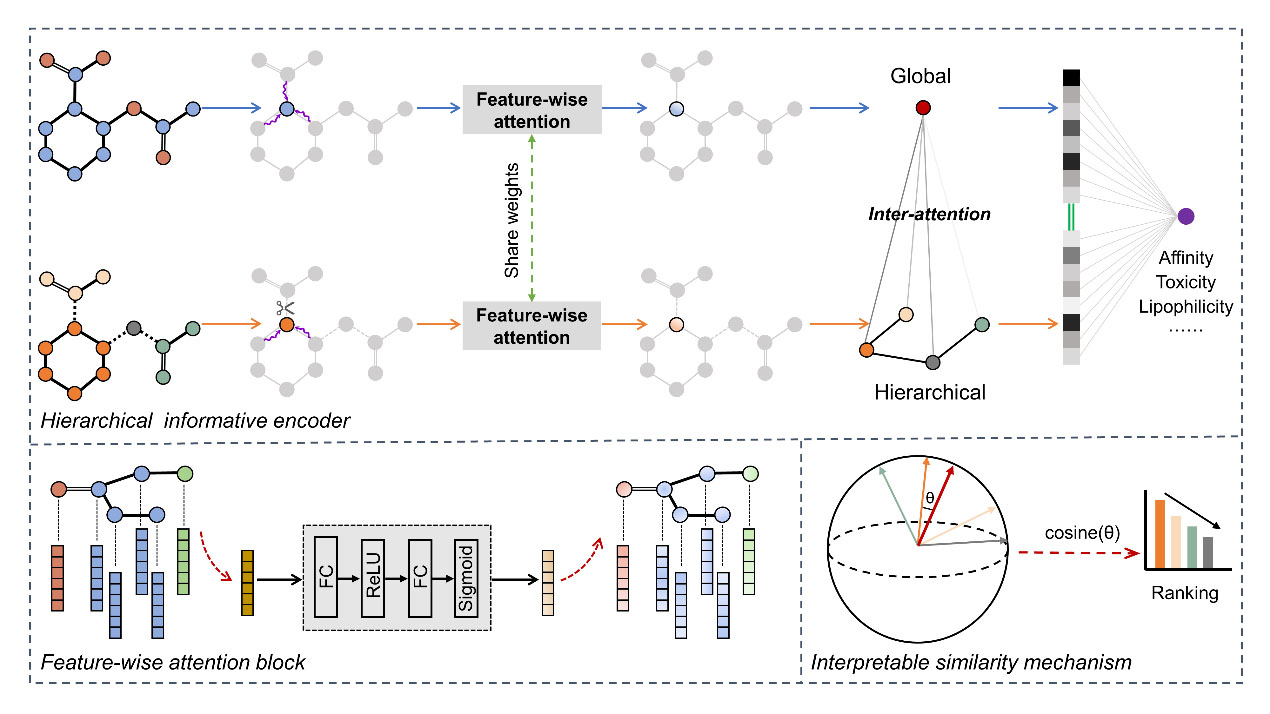
图1 HiGNN算法的总体框架
作者在涵盖物理化学、生物物理学、生理学性质和毒性的11个药物发现相关的基准数据集上系统地评估了HiGNN模型的性能。实验表明(表1),与所有基线模型相比,HiGNN模型在11个基准数据集的14个学习任务中的10个任务上实现了最先进的性能。此外,详细的消融研究表明(表2),BRICS信息和特征注意块都可以提高HiGNN模型的预测性能。
表1 HiGNN算法在11个基准数据集上的性能
Dataset | Split Type | Metric | Chemprop | GCN | GAT | Attentive FP | HRGCN+ | XGBoost | HiGNN |
BACE | random | ROC-AUC | 0.898 | 0.898 | 0.886 | 0.876 | 0.891 | 0.889 | 0.890 |
scaffold | ROC-AUC | 0.857 |
|
|
|
| 0.882 | ||
HIV | random | ROC-AUC | 0.827 | 0.834 | 0.826 | 0.822 | 0.824 | 0.816 | 0.816 |
scaffold | ROC-AUC | 0.794 |
|
| 0.802 | ||||
MUV | random | PRC-AUC | 0.053 | 0.061 | 0.057 | 0.038 | 0.082 | 0.068 | 0.186 |
Tox21 | random | ROC-AUC | 0.854 | 0.836 | 0.835 | 0.852 | 0.848 | 0.836 | 0.856 |
ToxCast | random | ROC-AUC | 0.764 | 0.770 | 0.768 | 0.794 | 0.793 | 0.774 | 0.781 |
BBBP | random | ROC-AUC | 0.917 | 0.903 | 0.898 | 0.887 | 0.926 | 0.926 | 0.932 |
scaffold | ROC-AUC | 0.886 |
|
| 0.927 | ||||
ClinTox | random | ROC-AUC | 0.897 | 0.895 | 0.888 | 0.904 | 0.899 | 0.911 | 0.930 |
SIDER | random | ROC-AUC | 0.658 | 0.634 | 0.627 | 0.623 | 0.641 | 0.642 | 0.651 |
FreeSolv | random | RMSE | 1.009 | 1.149 | 1.304 | 1.091 | 0.926 | 1.025 | 0.915 |
ESOL | random | RMSE | 0.587 | 0.708 | 0.658 | 0.587 | 0.563 | 0.582 | 0.532 |
Lipo | random | RMSE | 0.563 | 0.664 | 0.683 | 0.553 | 0.603 | 0.574 | 0.549 |
表2 HiGNN模型的消融实验
Method | BACE | BBBP | ClinTox | SIDER | Tox21 | FreeSolv | ESOL | Lipo |
HiGNN | 0.890 | 0.932 | 0.930 | 0.651 | 0.856 | 0.915 | 0.532 | 0.549 |
w/o HI | 0.887 | 0.930 | 0.926 | 0.654 | 0.852 | 0.941 | 0.536 | 0.575 |
w/o FA | 0.880 | 0.927 | 0.927 | 0.649 | 0.840 | 0.964 | 0.616 | 0.591 |
w/o All | 0.877 | 0.918 | 0.924 | 0.639 | 0.830 | 0.946 | 0.670 | 0.623 |
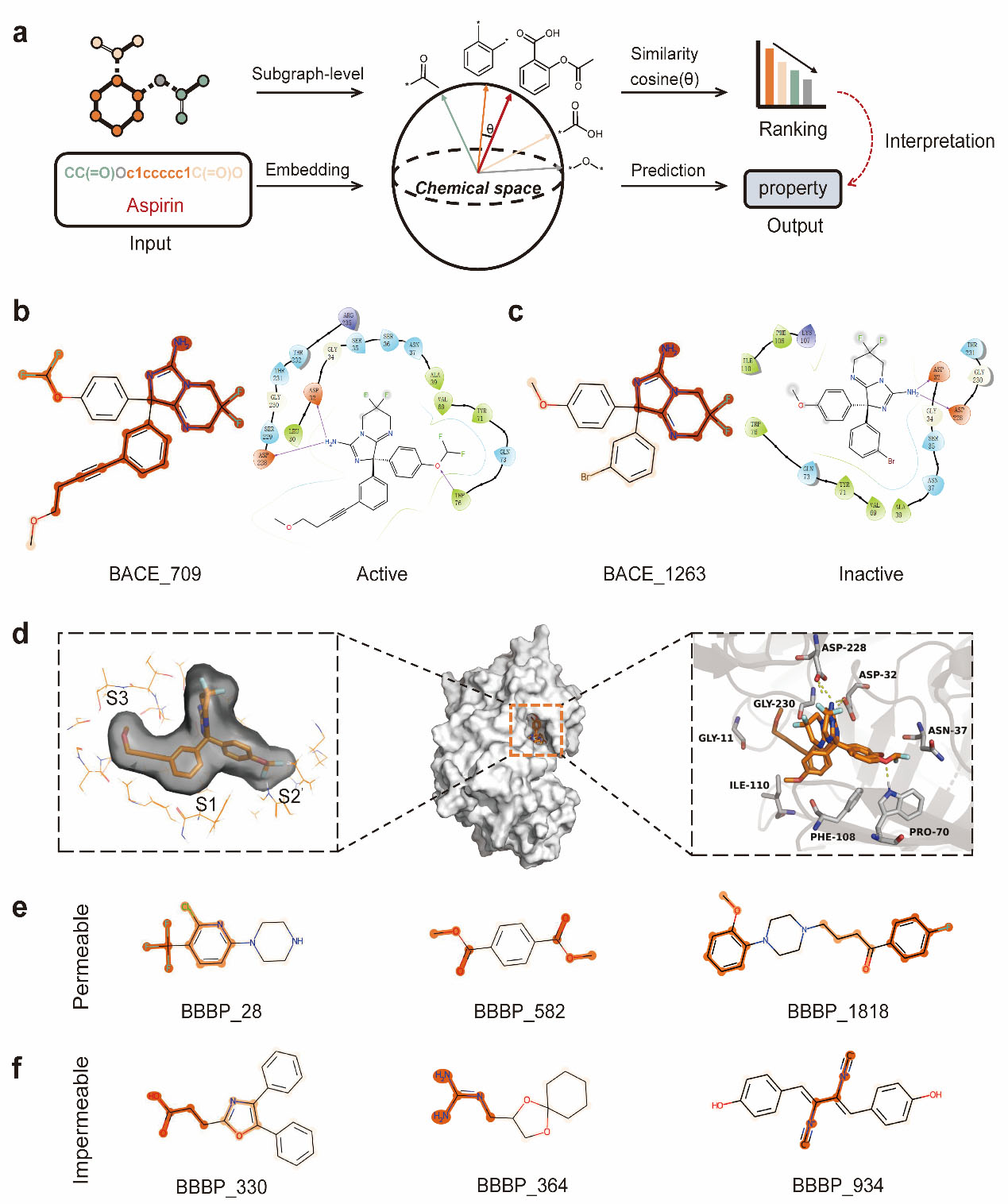
图2 HiGNN模型在BACE和BBBP数据集上的可解释性分析
最后,在案例分析中(图2),作者通过分子-片段相似性机制验证了HiGNN模型的可解释性。通过分子-片段相似性机制,HiGNN能识别出BACE以及BBBP数据集中分子的关键成分,从而为化学家提供了药物性质预测中具有重要价值的分子片段。
综合而言,相比当前优秀的机器学习和深度学习算法,该课题组开发的HiGNN深度学习算法其考虑了分子的层次信息和特征通道之间的关系,具有更优秀的预测性能。HiGNN首次提出了通用的特征注意块,作者希望该模块能够在GNN中进行更加广泛的应用。此外,HiGNN首次研究探索了在片段级别的可解释性,作者期望HiGNN作为强大的深度学习工具,可以帮助化学家识别分子中的关键结构,从而设计出具有预期特性或功能的分子。
上述研究结果得到国家自然科学基金面上项目(81973241)和广东省自然科学基金面上项目(2020A1515010548)的支持。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jcim.2c01099
代码链接:https://github.com/idrugLab/hignn