Science:像酶一样活化氧气,MOF稳定高自旋铁氧物种
2023-11-03 08:43·XMOL学术
导读
在自然界中,非血红素铁酶使用氧气生成高自旋四价铁氧物种,用于各种氧化反应。几十年来,尽管合成化学家进行了很多尝试,但是在人造系统中模拟这一过程——像酶一样活化氧气生成高自旋四价铁氧物种——仍是一个尚未实现的目标。加州大学伯克利分校化学系的Jeffrey R. Long教授领导的团队报道了一种新型的金属-有机框架(MOF)材料,其中含有的二价铁位点局部结构与α-酮戊二酸依赖性双加氧酶(ɑ-ketoglutarate-dependent dioxygenases)的活性中心类似。该材料在低温(低至100 K)下可以与氧气直接反应,生成高自旋四价铁氧物种。作者通过多种谱学方法对该铁氧物种进行了表征,包括原位漫反射红外光谱(in situ diffuse reflectance Fourier transform, DRIFTS)、原位和可变场穆斯堡尔谱(Mossbauer spectroscopy)、Fe Kβ X射线发射光谱(X-ray emission spectroscopy, XES)和核共振振动光谱(Nuclear resonance vibrational spectroscopy, NRVS)。实验结果表明,在氧气环境中,该MOF材料能够催化环己烷的氧化和乙烷到乙醇的化学计量转化。相关研究成果于2023年11月3日在线发表于《科学》(Science)杂志。
如何开发使用氧气进行碳氢化合物选择性氧化的催化剂,是全球发展绿色化学技术的重要挑战之一。自然界中,单核非血红素铁金属酶利用氧气进行C-H键氧化反应,例如普遍存在的α-酮戊二酸依赖性双加氧酶。其中研究最多的酶之一是牛磺酸-α-酮戊二酸依赖性双加氧酶(TauD),它可以氧化牛磺酸α位的一个C-H键。TauD及其一系列双加氧酶的反应性的关键是高自旋(S = 2)Fe(IV)=O中间体,该中间体在二价铁与氧气耦合以及α-酮戊二酸的氧化和脱羧之后形成(图1A)。在过去的几十年里,为了更好地理解和模仿生物系统中的反应性,研究者投入了大量精力以合成与分析分子化合物和铁-沸石模型体系中的四价铁氧物种。但是迄今为止,大多数研究的铁氧物种都具有中间的自旋基态(S = 1),而且只有少数是在溶液中使用氧气作为氧化剂而生成的。高自旋Fe(IV)=O化合物只能通过氧化剂如三甲基铵-N-氧化物、高价碘试剂,或者氧气在光照下裂解O-O键的方式来获得。像天然酶一样仅仅使用氧气来生成高自旋Fe(IV)=O物种并实现与天然酶类似的反应性仍是一个不小的难题。
近年来,MOF被视为一种适用于仿生化学研究的材料。由于具有多孔和结构可调的性质,MOF材料还适合用来探索固体-气体反应中的反应机理。将金属中心固定在晶格中,这有助于防止高自旋四价铁氧中间体通过二聚化或分子内配体氧化而分解。于在此背景下,Fe1.5Zn3.5Cl4(btdd)3(H2btdd = bis(1H-1, 2, 3-triazolo[4, 5-b], [4', 5'-i])dibenzo[1,4]dioxin)作为潜在的仿生材料脱颖而出。MOF材料FexZn5-x(prv)4(btdd)3(x = 1 or 1.8; Hprv = 丙酮酸)和FeZn4(moba)4(btdd)3(Hmoba = 3,3-二甲基-2-羟基丁酸)可与氧气反应生成高自旋(S = 2)的Fe(IV)=O化合物,并可以氧化碳氢化合物(图1B和1C)。
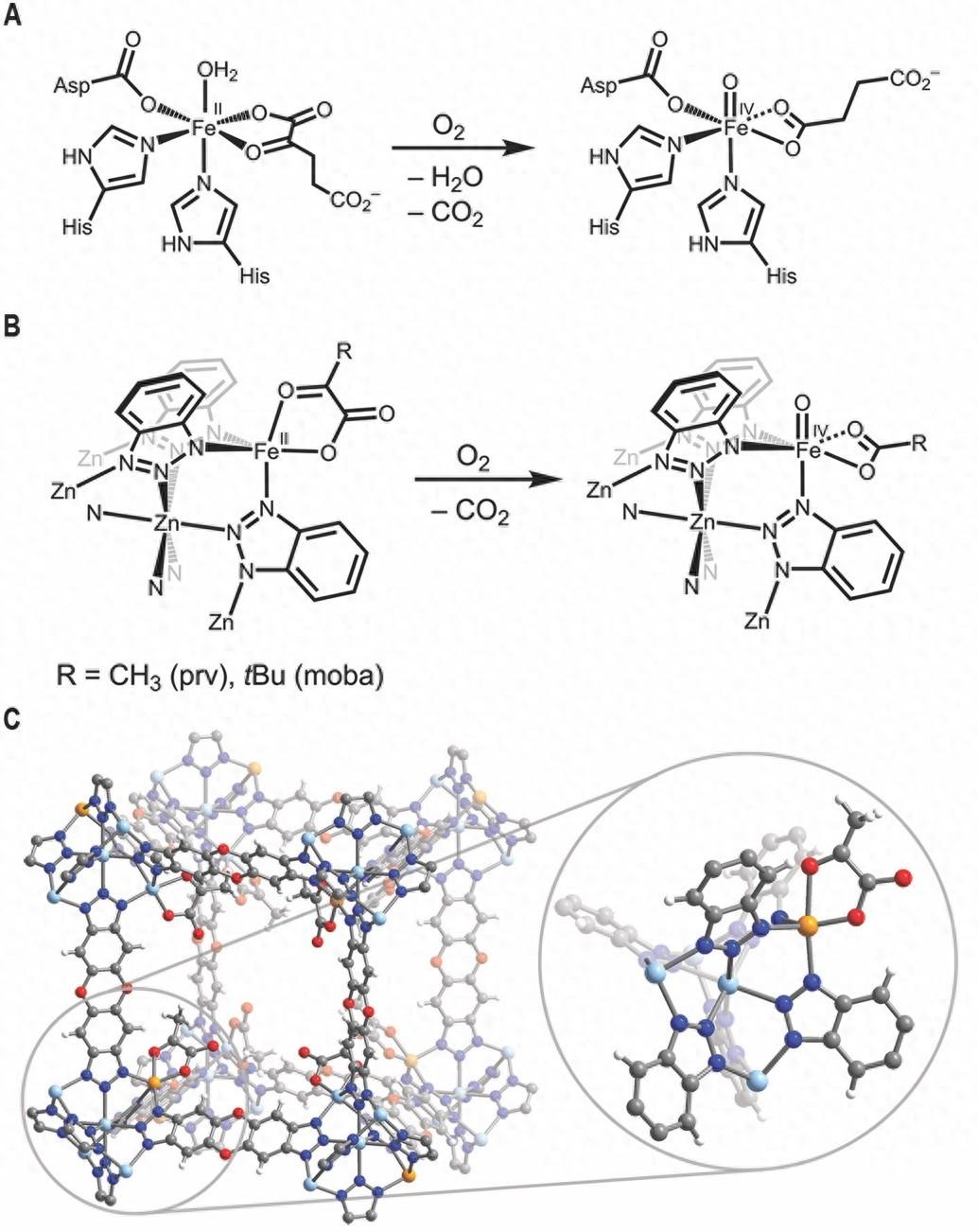
图1. α-酮戊二酸依赖性双加氧酶和MOF材料中反应机理的比较。图片来源:Science
含铁的MOF由经典的离子交换方法合成。将Zn5Cl4(btdd)3 (MFU-4l) 浸泡在二氯化铁溶液中,得到FexZn5−xCl4(btdd)3材料,再使用阴离子交换,将氯离子置换为丙酮酸根离子。在此材料中,铁金属中心的配位环境与TauD活性中心相似(图1B,C)。相关表征可以阅读原文。作者使用原位DRIFTS光谱成功观察到在Fe(IV)=O的形成。低温环境下(100 K),活化后的Fe1.8Zn3.2(prv)4(btdd)3在氧气环境中,DRIFTS光谱中逐渐出现了一个在2341 cm−1的新吸收峰,对应为吸附在MOF上的CO2气体分子。当利用18O2 和13C-丙酮酸进行双标记实验时,CO2吸收带转变为2275 cm−1,对应到13CO2的形成而非13C18O2,说明配位在铁中心的丙酮酸进行了脱羧反应,CO2中的氧原子完全来自于丙酮酸而非O2。在100 K,向Fe1.8Zn3.2(prv)4(btdd)3通入氧气并观察时,作者观察到一个吸收峰在831 cm−1处逐渐增强,他们认为这是Fe(IV)=O伸缩振动。随着温度升高到200 K,这个吸收峰的强度增加,但在250 K时显著减弱,并在298 K时消失。当使用18O2作为氧化剂时,吸收峰移动到了796 cm−1,这与使用简单谐振子模型计算得到的Fe(IV)=18O的伸缩频率794 cm−1相符。并且,这于氧化酶TauD-J 中的Fe(IV)=O和Fe(IV)=18O 红外吸收峰十分接近(分别在821 and 787 cm−1)。DRIFTS结果强有力地证明了200 K下Fe(IV)=O的形成。同时,作者观察到,将温度上升到250 K 后Fe(IV)=O的红外信号强度开始降低,进一步上升到室温后,Fe(IV)=O信号完全消失。作者认为,随着温度升高,Fe(IV)=O开始分解并形成了Fe(III)–OH的结构。
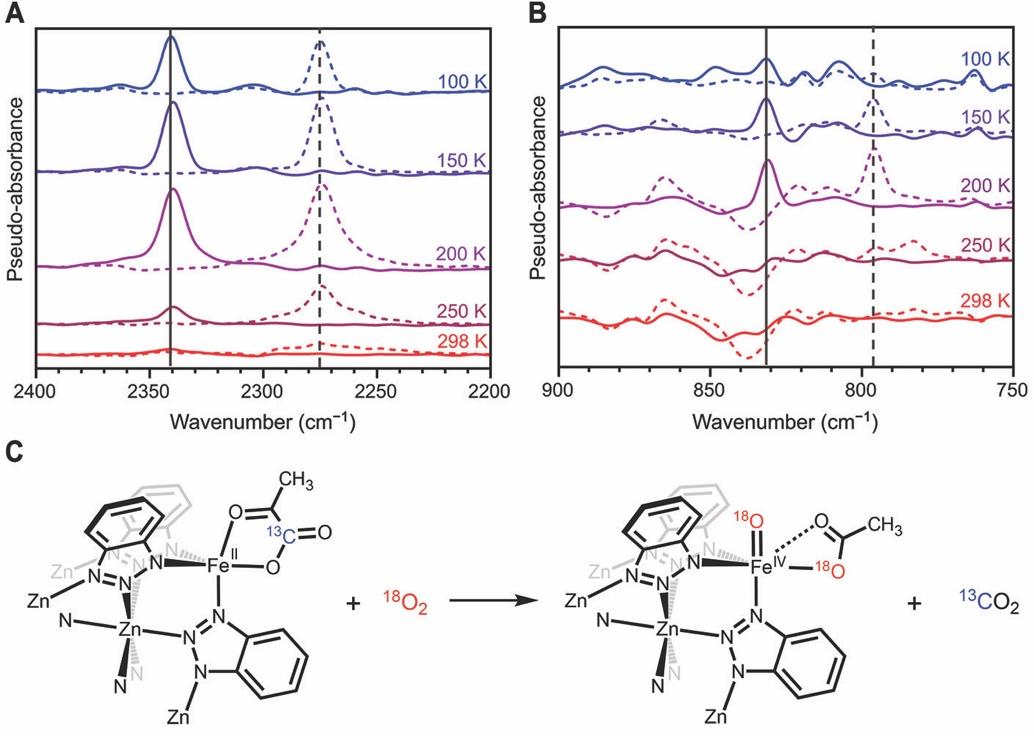
图2. 高自旋四价铁氧物种的原位漫反射红外光谱分析。图片来源:Science
作者进一步通过原位穆斯堡尔光谱对FexZn5−x(prv)4(btdd)3与氧气反应生成的物种进行了研究。在5 K温度下,FeZn4(prv)4(btdd)3的穆斯堡尔光谱显示了一个主要的双峰,表明存在高自旋、五配位的铁(II)位点。在100 K下通入300 mbar的氧气后,这个双峰保持不变,但是出现了一个新的双峰,其同质异能位移δ = 0.260(4) mm/s,四极分裂|∆EQ| = 0.572(8) mm/s(占16.7(2)%的面积),作者认为这是四价铁氧物种。这些参数与TauD的S = 2 Fe(IV)=O中间体的参数相似(δ = 0.31 mm/s和∆EQ = −0.88 mm/s)。与其他报道的四价铁氧物种的δ和|∆EQ|进行详细比较,作者发现FeZn4(prv)4(btdd)3与氧气反应生成的铁氧物种与S = 2自旋态(δS=2 = 0.02–0.37 mm/s; |∆EQ|S=2 = 0.23–1.27 mm/s)更为一致,而不是S = 1自旋态(δS=1 < 0.20 mm/s; |∆EQ|S=1 = 0.44–2.09 mm/s)。密度泛函理论(DFT)计算在B3LYP/def2-TZVP理论水平下预测了一个S = 2的六配位Fe(IV)=O结构的δ为0.25 mm/s,与实验值吻合。
当在125 K和150 K下,向FeZn4(prv)4(btdd)3继续通入氧气,Fe(IV)=O双峰的面积百分比可以增加到最大的20.0(2)%。然而,在163 K通入氧气并未导致Fe(IV)=O双峰的百分比增加。在此温度下,有可能最初生成了更多的Fe(IV)=O物种,但其中一些位点与乙酸配体反应,生成一个羟基化产物并重新形成高自旋铁(II)位点。类似的分子内α-C–H键羟基化在α-酮戊二酸依赖型双氧酶中此前有过报道。最后,在没有额外通入氧气的情况下,在250 K和298 K加热样品后,收集了5 K下的光谱。在250 K加热后,Fe(IV)=O双峰的面积只有9.7(3)%;在298 K加热后,Fe(IV)=O双峰消失,这与DRIFTS数据中观察到的分解现象一致。
通过对FeZn4(prv)4(btdd)3中生成的Fe(IV)=O物种的同质异能位移(δ)和四极分裂(|∆EQ|)值的测定,作者发现这些参数更符合S = 2的自旋状态,而不是S = 1,但仅凭这些参数并不足以准确判断Fe(IV)=O的自旋状态。为了从实验上更为准确地确认自旋状态,作者转向应用变场穆斯堡尔谱学。作者预期,由于氧气氧化Zn4(prv)4(btdd)3仅可达到的Fe(IV)=O物种的低比例,数据分析将受到限制。因此,作者合成了FeZn4(moba)4(btdd)3,其特点是带有α位有叔丁基团的3,3-二甲基-2-酮丁酸根配体。这种材料与FeZn4(prv)4(btdd)3具有类似的结构。用氧气在100 K氧化FeZn4(moba)4(btdd)3后,原位DRIFTS数据支持通过3,3-二甲基-2-酮丁酸的脱羧作用生成Fe(IV)=O物种(ν(Fe=O) = 828 cm−1, ν(Fe=18O) = 794 cm−1),这与上面讨论的FeZn4(prv)4(btdd)3的反应性一致。
零场穆斯堡尔谱的FeZn4(moba)4(btdd)3在5 K时显示了一个主要的双峰,归属于高自旋、五配位的铁(II)位点。经过一系列在低温下的氧气原位氧化后,5 K 穆斯堡尔谱中出现了一个新的双峰,其δ = 0.292(1) mm/s,∆EQ = −0.603(1) mm/s(占总面积的61.7(1)%),归属于Fe(IV)=O物种。这个相对面积显著大于原位被氧气氧化的FeZn4(prv)4(btdd)3的最大相对面积。随后,作者收集了被氧气氧化的FeZn4(moba)4(btdd)3材料的变场穆斯堡尔数据:应用的磁场强度从0 T到7 T不等,同时在7 T的磁场下,温度从1.7 K到40 K变化。他们还收集了在低于5 K的温度和从0到7 T的磁场范围内的FeZn4(moba)4(btdd)3的谱图,以获取用于模拟被氧气氧化的材料残余铁(II)物种的固定参数。同时对变场和变温谱的建模是通过对四个子谱的自旋哈密顿量进行对角化来完成的,以获得无耦合Fe(IV)=O物种的零场分裂(D)和超精细耦合参数(Axx, Ayy, Azz)。基于与已报告的Fe(IV)=O物种的比较,虽然数据可以用S = 2或S = 1模型来拟合,但只有S = 2模型的结果在化学上是合理的。对于S = 2 Fe(IV)=O物种的最佳模型给出了D = 12.7(6) cm−1和Aiso = −16.4 T,这些参数与S = 2 Fe(IV)=O物种的报告值极好地一致(DS=2 = 4–14 cm−1, Aiso(S=2) = −23 to −16 T)。相比之下,S = 1 Fe(IV)=O位点的最佳模型给出了与先前为S = 1 Fe(IV)=O物种报告的值显著不同的结果。
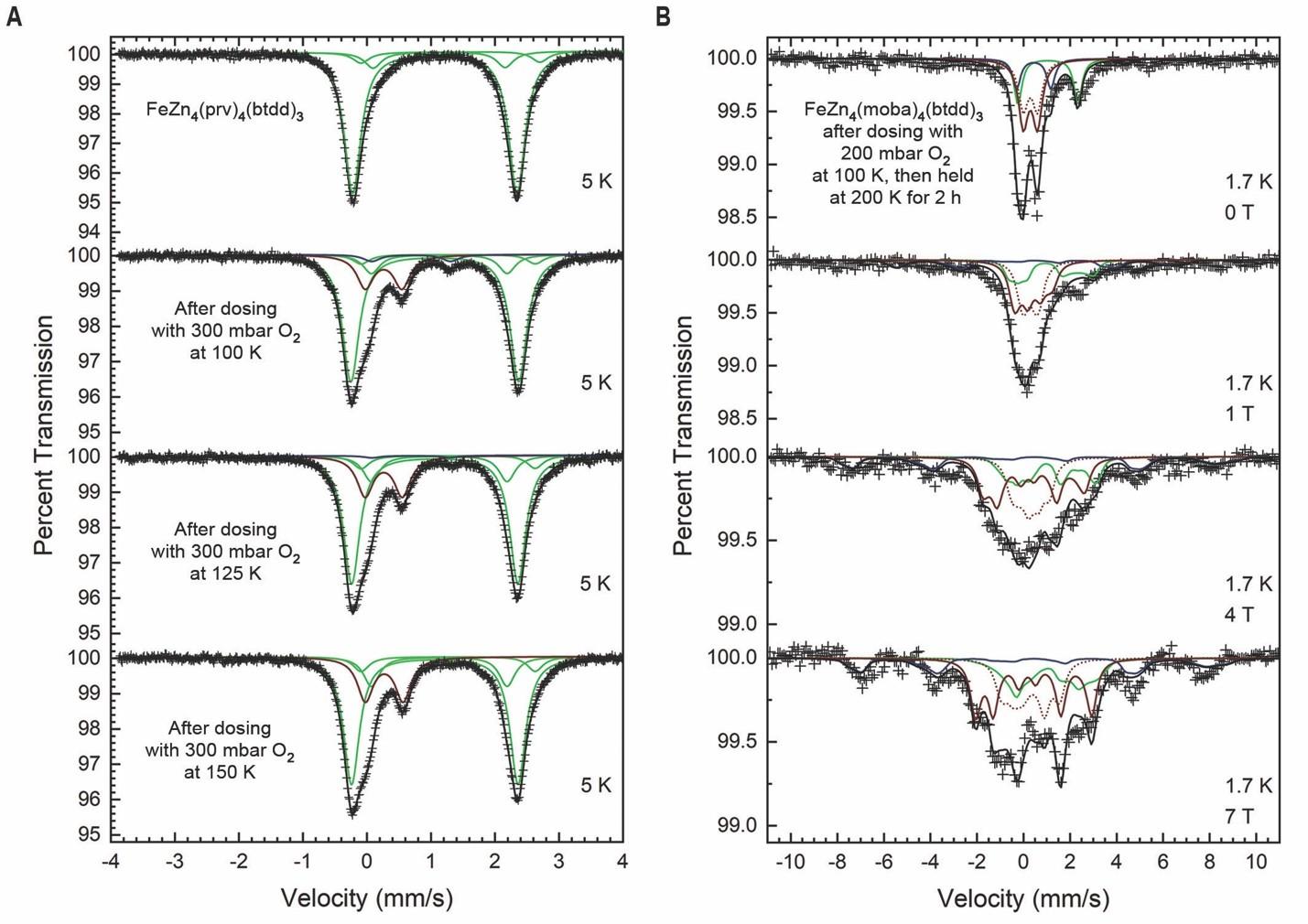
图3. 高自旋四价铁氧物种的原位和可变场穆斯堡尔谱分析。图片来源:Science
作者还使用了核共振振动光谱(NRVS)来进一步了解Fe(IV)=O物种的局部结构。这种表征技术可以选择性地获得穆斯堡尔活性核的完整振动模式集,因此能够提供其他光谱方法无法获得的结构信息。图4中,氧气通入样品后,新出现的822 cm−1峰被指定为Fe(IV)=O的振动,其大小与其他合成系统中报告的非血红素Fe(IV)=O物种相似。当使用18O2进行实验时,振动转而出现在788 cm−1处。当检测结束后,将同样的材料加热至298 K后,原来被认为是Fe(IV)=O峰消失。显示该Fe(IV)=O物种在较高温度下会分解,这于先前红外和穆斯堡尔谱得到的结果相吻合。
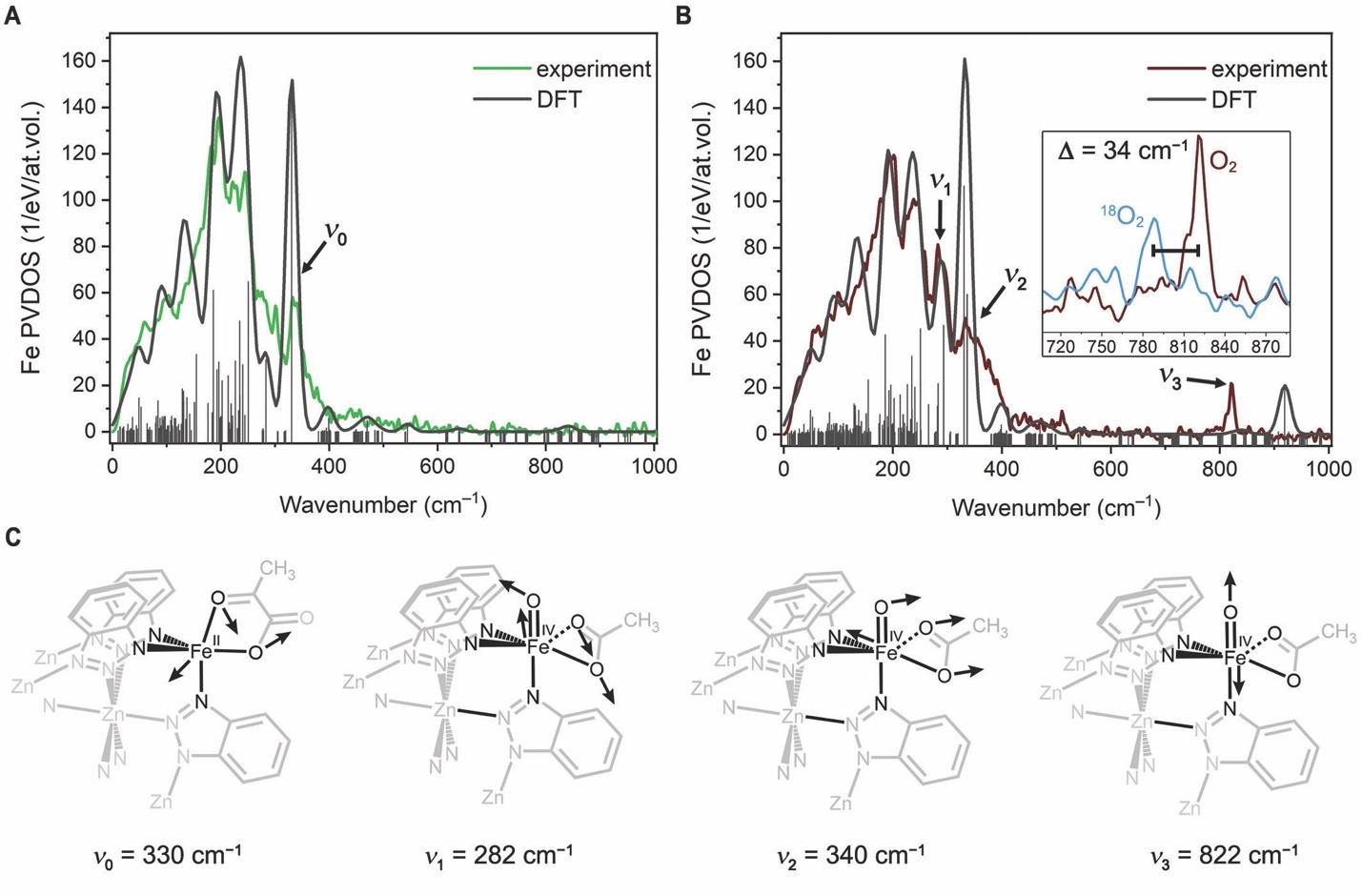
图4. 高自旋四价铁氧物种核共振振动光谱分析。图片来源:Science
当使用氧气做氧化剂时,FeZn4(prv)4(btdd)3 可以氧化碳氢化合物。在21 °C下,作者将活化的FeZn4(prv)4(btdd)3浸泡在环己烷中,并通入1 bar的氧气。24小时后,1H 核磁共振波谱和GC-MS显示产物为环己醇(NMR产率为22%),没有检测到环己酮。之后,作者在原反应条件中,加入了过量丙酮酸促进催化周转。在该反应条件下观察到产物为环己酮和环己醇,其比例为2:1,总产率为173%。作者将乙烷和氧气的高压混合气体通入含有Fe1.8Zn3.2(prv)4(btdd)3的高压反应釜中,产物为乙醇和乙醛,其比例为3:1,总产率高达82%。
总结
作者报道了MOF材料FexZn5−x(prv)4(btdd)3和FeZn4(moba)4(btdd)3,它们在100 K下能够通过铁(II)位点活化氧气,生成高自旋四价铁氧物种。这种反应性类似TauD中氧气被活化过程。这些人工合成MOF材料能够很好的模拟自然界的酶,活化氧气生成高自旋Fe(IV)=O中间体,催化碳氢化合物的氧化反应,包括将乙烷氧化为乙醇。这项工作为开发含铁MOF作为模拟金属酶反应性的异质催化剂奠定了基础。
Reactive high-spin iron(IV)-oxo sites through dioxygen activation in a metal–organic framework
Kaipeng Hou†, Jonas Börgel†, Henry Z. H. Jiang, Daniel J. SantaLucia, Hyunchul Kwon, Hao Zhuang, Khetpakorn Chakarawet, Rachel C. Rohde, Jordan W. Taylor, Chaochao Dun, Maria V. Paley, Ari B. Turkiewicz, Jesse G. Park, Haiyan Mao, Ziting Zhu, E. Ercan Alp, Jiyong Zhao, Michael Y. Hu, Barbara Lavina, Sergey Peredkov, Xudong Lv, Julia Oktawiec, Katie R. Meihaus, Dimitrios A. Pantazis, Marco Vandone, Valentina Colombo, Eckhard Bill, Jeffrey J. Urban, R. David Britt, Fernande Grandjean, Gary J. Long, Serena DeBeer, Frank Neese, Jeffrey A. Reimer, Jeffrey R. Long
Science, 2023, 382, 547-553, DOI: 10.1126/science.add7417