海水也能产H₂O₂!单原子光催化最新JACS!
原创2023-07-29 10:30·邃瞳科学云


第一作者:Peng Ren
通讯作者:Shoubhik Das
通讯单位:比利时安特卫普大学
论文DOI:
https://doi.org/10.1021/jacs.3c03785
全文速览
本文开发了一种芳基氨取代的石墨氮化碳(g-C3N4)催化剂,其具有原子级分散的Mn,能够直接从海水中产生过氧化氢(H2O2)。这种新型催化剂表现出优异的反应活性,在相同条件下,可以在7 h内从碱性水中获得达2230μM的H2O2,从海水中获得高达1800μM的H2O2。更重要的是,催化剂可以快速回收以供后续使用,而性能没有明显损失。值得注意的是,与通常的双电子氧还原反应途径不同,H2O2是通过不常见的双电子水氧化反应(WOR)过程生成的。其中,直接和间接WOR过程都会发生,即光诱导h+通过一步2e-WOR直接将H2O氧化为H2O2,或者,光诱导h+首先氧化氢氧根(OH-)离子生成羟基自由基(•OH),两个•OH间接结合形成H2O2。
背景介绍
在研究单原子催化剂(SAC)方面,将金属分散到特定载体(如沸石、MOF、COF、碳载体、氧化物和碳氮化物等)上可以增强孤立分散金属原子的使用,并提供更多的活性位点。实践证明,SACs可以增强特定反应的催化反应活性和选择性。虽然 Fe、Ni、Ru 和 Pt 等金属作为 SAC 已得到广泛研究,但其他 3d 过渡金属(从 Sc 到 Mn)由于其反应性较低,仍较少被研究。
H2O2作为高效绿色氧化剂被广泛应用于有机工业合成、纸浆和造纸漂白工业、污水处理以及消毒剂等领域,并可直接用于单室燃料电池发电,其能量密度与 H2 相当,且更容易储存和运输。由于全球市场对H2O2的需求不断增强,通过水的光催化氧化转化人工合成H2O2被视为一种非常有前途的替代方案。人们报道了不同的均质光催化剂、团簇、纳米粒子、和 SAC 基材料,例如, Sb 和 Pd分别以原子方式分散在氮化碳和剥离石墨碳氮化物上。SAC 基光催化剂遵循双电子氧还原反应 (ORR) 途径,其中间体 μ-过氧化物的形成发生在单原子位点。在大多数情况下,牺牲质子供体(例如乙醇、2-丙醇和苯甲醇)对于加速电子空穴对的分离以提高反应性至关重要。相比之下,SAC 的水氧化反应 (WOR) 很少被研究,其中光致空穴 (h+) 通过一步 2e– WOR 直接将 H2O 氧化为 H2O2,或氧化 H2O生成羟基自由基 (·OH),再由羟基自由基 (·OH) 重新结合形成 H2O2。值得一提的是,地球上全部水的97%是海水,只有3%是淡水。海水中各种盐的存在常常导致光催化剂失活,从而限制了其实际应用。虽然电催化方面已经取得了一些进展,但光催化方法仍处于起步阶段。
图文解析
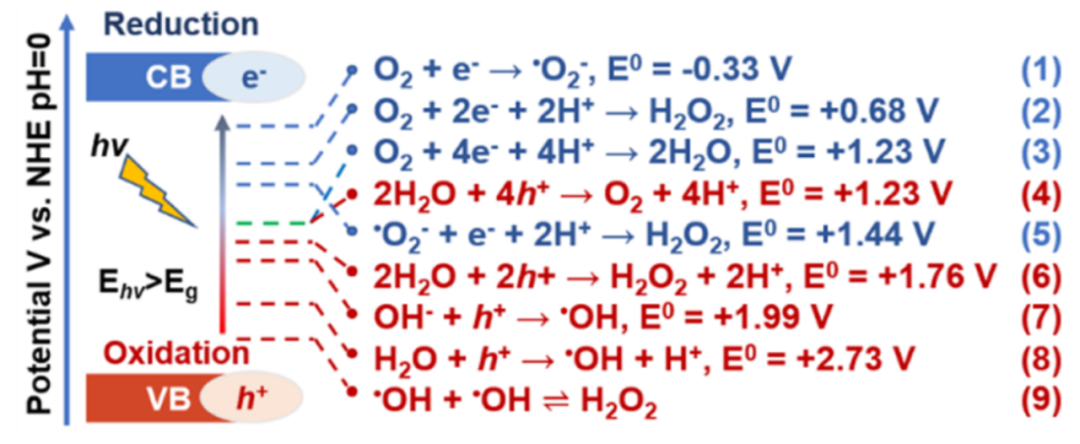
图1. 光催化 O2 还原和 H2O氧化为 H2O2 的能量图,显示了相关的标准势。
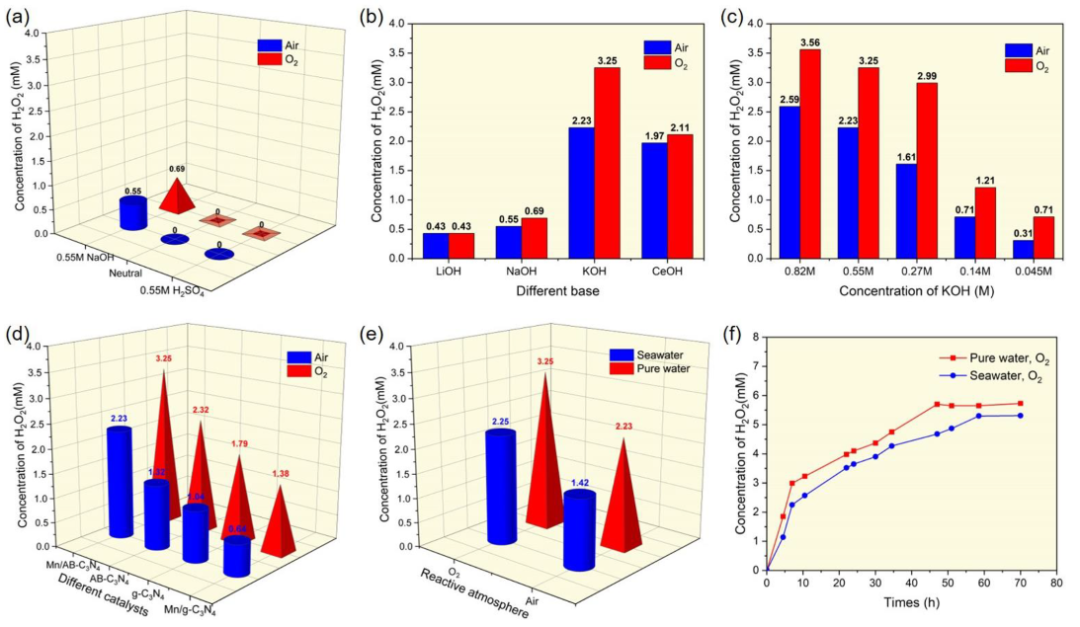
图2. (a) 在不同 pH 介质下使用 Mn/AB-C3N4催化剂光催化生产 H2O2。(b) 使用 Mn/AB-C3N4 催化剂与各种碱金属氢氧化物光催化生产 H2O2。(c) 使用具有不同浓度 KOH 的 Mn/AB-C3N4 催化剂光催化生产 H2O2。(d) 使用其他改性 g-C3N4 催化剂光催化生产 H2O2。(e) 在不同反应气氛下去离子水和海水的比较。(f) 在去离子水和海水中光催化产生H2O2的动力学实验。
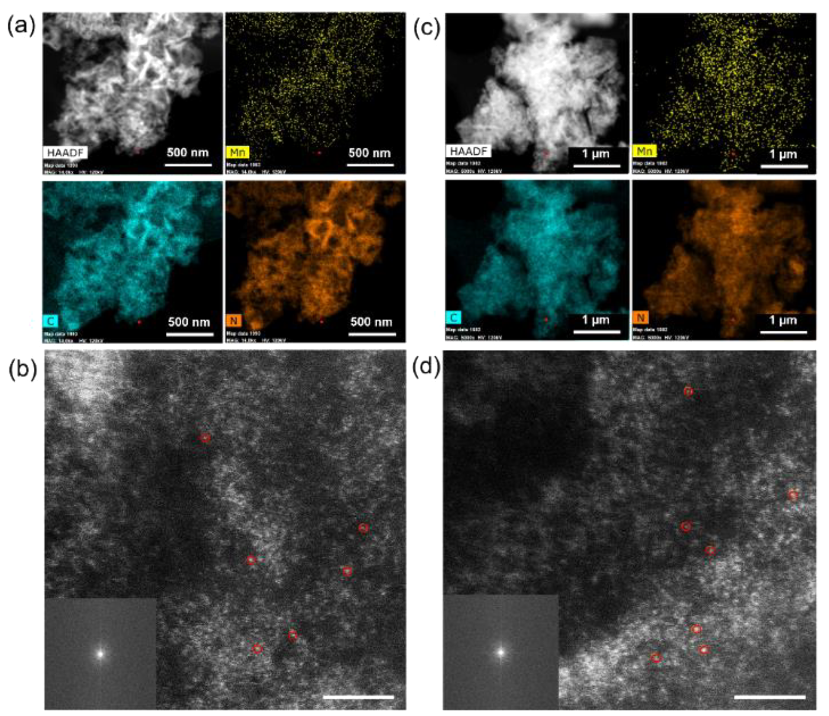
图3. 反应前:(a) 低倍 STEM-EDS 元素图,显示催化剂上存在 Mn、C 和 N(比例尺:500 nm)。(b) 分散在催化剂上的原子级 Mn 位点的 HAADF-STEM 图像(比例尺:2 nm)。反应后:(c) 低倍STEM-EDS 元素图,显示催化剂上存在 Mn、C 和 N(比例尺:1 μm)。(d) HAADF-STEM 图像,显示分散在 C3N4 上的 Mn 原子级位点(比例尺:2 nm)。
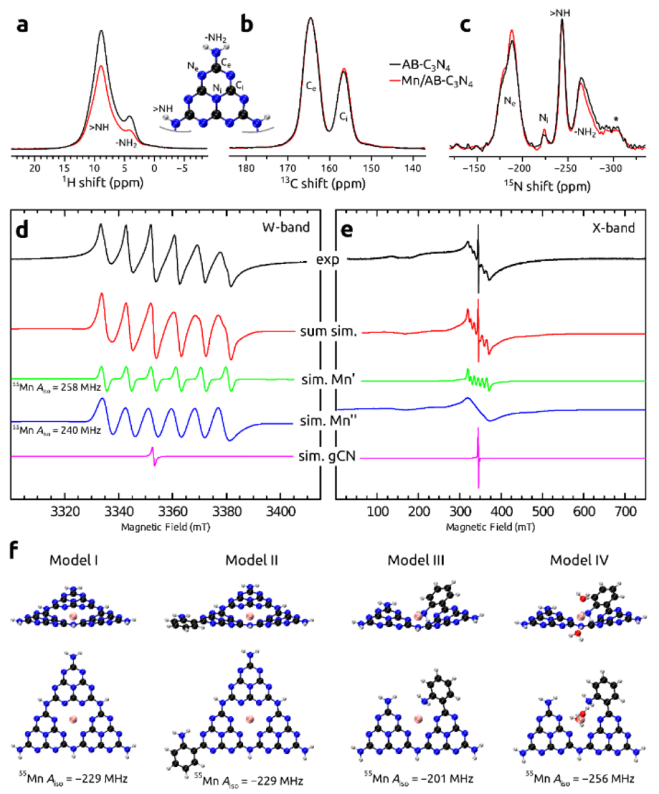
图4. AB-C3N4催化剂和Mn/AB-C3N4催化剂的 (a) 1H MAS、(b) 13C CPMAS和 (c)15N CPMAS NMR谱。(d) Mn/AB-C3N4催化剂的连续 W 波段和 (e) 连续 X 波段 EPR 谱,以及各自的EPR 信号解卷积。(f) 催化剂中顺磁性 Mn2+离子的环境模型和计算的 55Mn 超精细耦合常数。
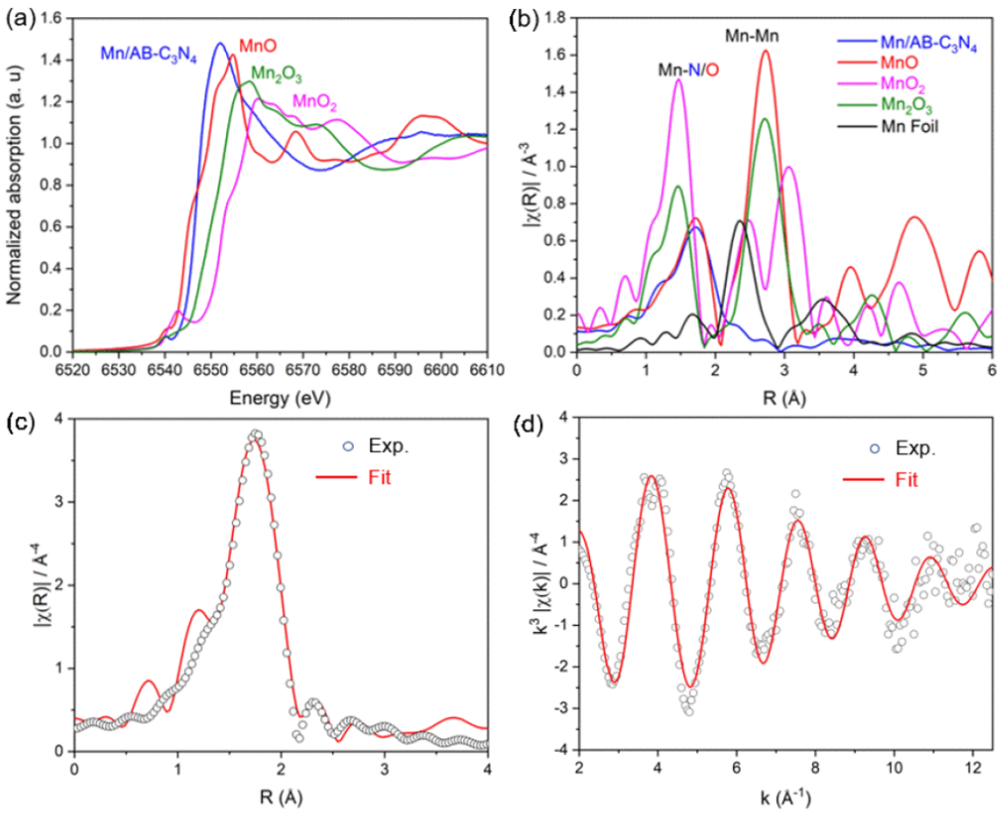
图5. Mn/AB-C3N4 光催化剂、MnO、Mn2O3、MnO2 和 Mn 箔的归一化 Mn K-edge XANES 光谱 (a) 和 k3 加权 EXAFS 的傅里叶变换 (b)。k 空间中 FT 信号 (c) 和 k3 加权 EXAFS 信号 (d) 的原始数据(空心圆圈)和拟合数据(红线)。
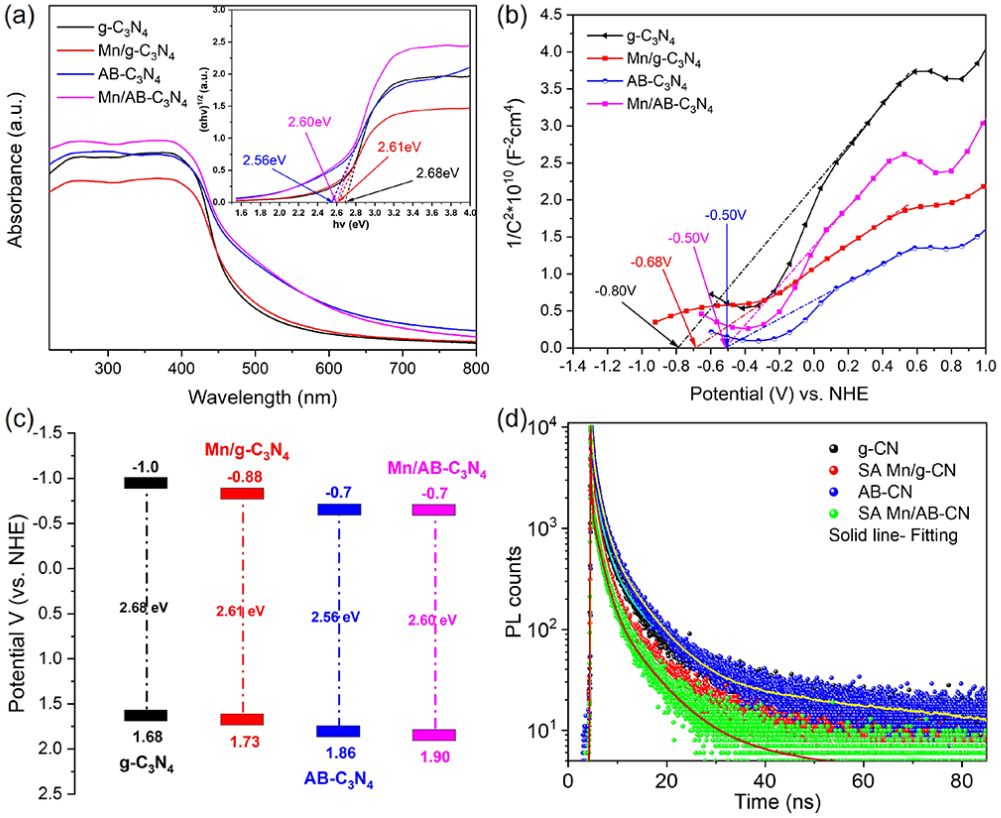
图6. (a) 块状 g-C3N4 及其衍生物的紫外可见吸收光谱和(插图)相应的紫外可见 DRS Tauc 图。(b) 块状g-C3N4 及其衍生物的莫特-肖特基图,揭示了在 1 kHz 频率下收集的平带电势值。(c) g-C3N4及其衍生物的电子带位置。(d) g-C3N4、SA Mn/g-C3N4、AB-C3N4和 SA Mn/AB-C3N4 粉末的 PL 寿命光谱。实线显示了衰减曲线的拟合。
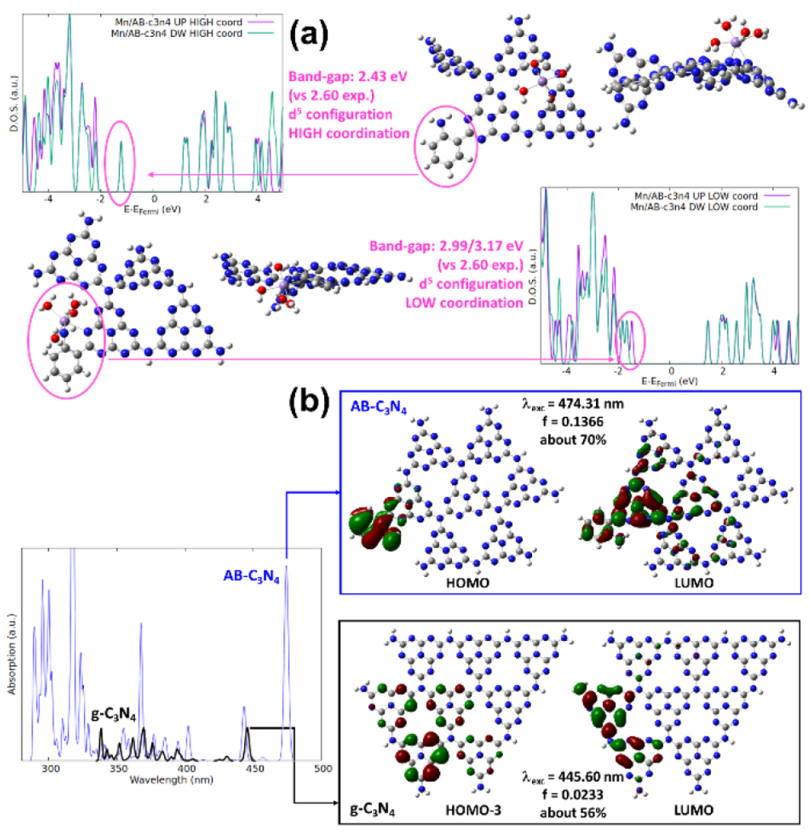
图7. (a) 在 DFT 水平上优化的两个系统的原子结构模型和态密度(DOS),其中 Mn2+ 位于高配位位点(上)和低配位位点(下);(b) g-C3N4(粗黑线)和 AB-C3N4(细蓝线)的 TDDFT 光谱,其中箭头指示了涉及主要跃迁的分子轨道。
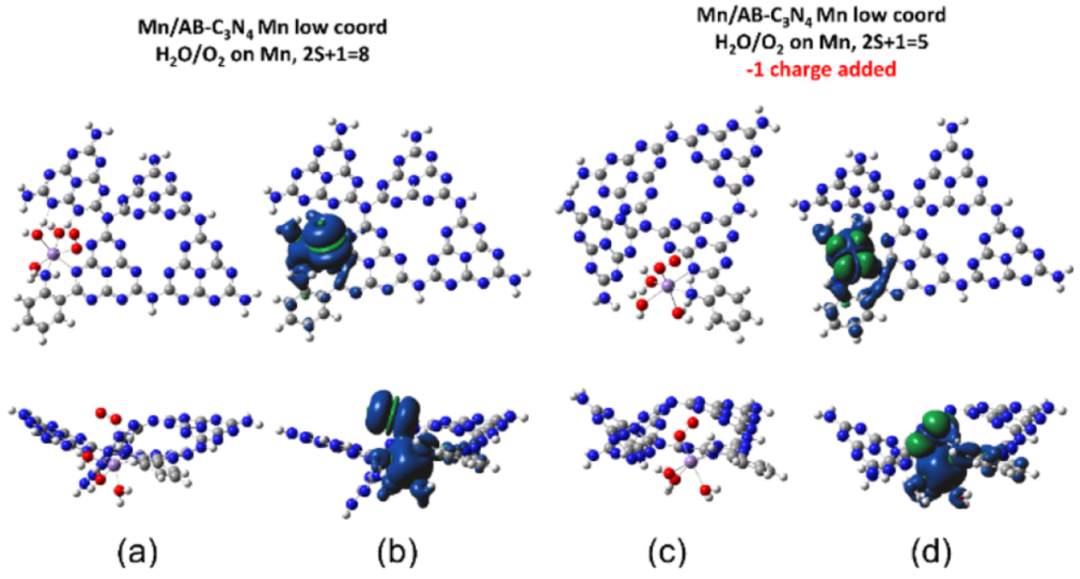
图8. 低配位位点Mn2+中心的O2吸附模型的(a) 俯视图、侧视图 和 (b) 自旋密度等值面;(c, d) 当向 (a, b) 的系统中添加额外电子时获得的对应图。颜色:N 蓝色、C 灰色、H 白色、O 红色和 Mn 紫色。
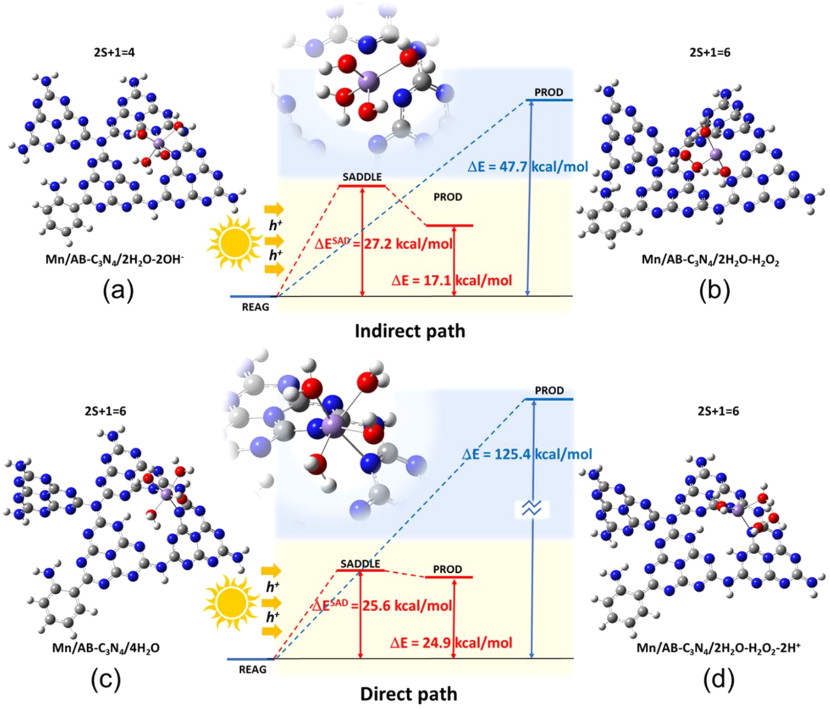
图9. (a) 2(OH)−+ 2H2O 和 (b) 2H2O + H2O2 的结构;(c) 4H2O 和 (d) 2H2O + H2O2 + 2H+的结构吸附在 Mn/AB-C3N4 载体的高配位 Mn2+ 上。颜色:N 蓝色、C 灰色、H 白色、O 红色和 Mn 紫色。
总结与展望
总的来说,该研究开发了一种锰基催化剂,其配位不饱和 Mn-Nx 位点原子级分散在芳基氨取代的石墨碳氮化物上。综合表征表明,g-C3N4 骨架上的 Mn 物种以孤立的 Mn 原子形式存在,平均与 4.6 个N/O 原子配位。值得注意的是,这种 Mn-SAPC 在不使用任何有机电子给体的情况下,能够从海水光催化生产 H2O2 ,并表现出优异的催化反应活性和稳定性。此外,实验结果和理论计算揭示了由单个Mn原子和三嗪环边缘的相邻氮原子组成的不饱和Mn-Nx位点与接枝到g-C3N4骨架上的芳基氨协同作用。并且芳基氨的引入缩短了带隙并提高了光吸收。Mn-Nx 位点促进了 O2 的吸附和活化,从而促进了光生电子空穴对的分离并抑制了复合,实现了出色的催化反应活性。总之,所报道的合成方法提供了一种从非贵金属过渡金属合成具有高度原子分散性和配位不饱和金属-Nx位点的金属-g-C3N4催化剂的替代方法,可进一步应用于其他(光)电催化反应和有机转化。