
【论文亮点】
阐明了多溴联苯醚光解的多步反应过程。
结合降解/生成动力学和计算化学提出并验证了多溴二苯并呋喃生成的自由基竞争反应机制。
证明了溶液中的氢供体是多溴二苯并呋喃生成的关键控制因素。
评估了不同多溴联苯醚在水溶液中生成多溴二苯并呋喃的潜力。
【图文摘要】
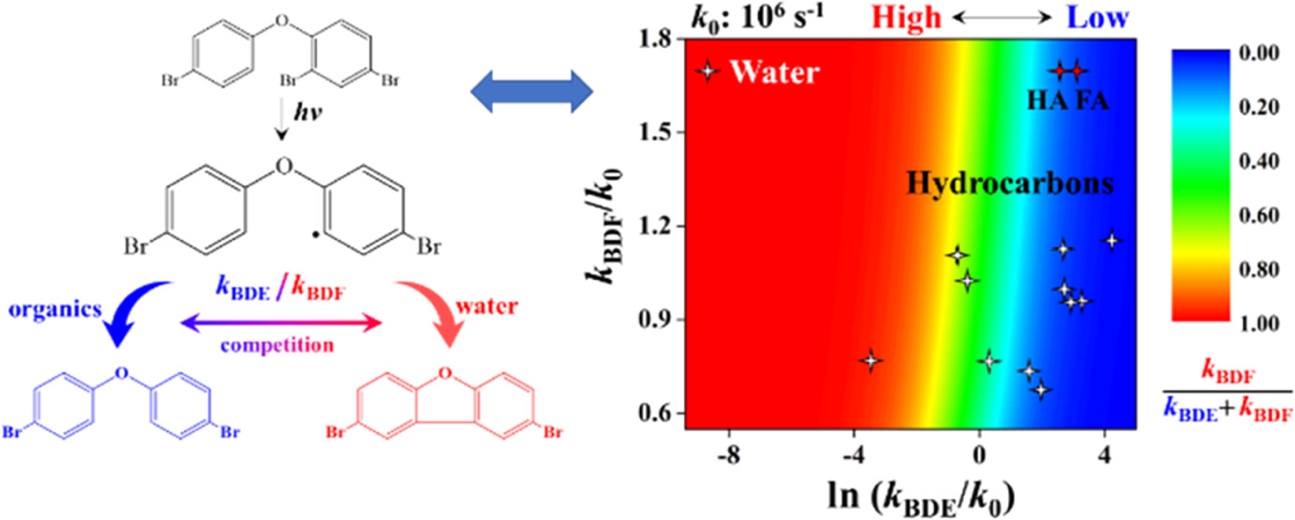
多溴二苯并呋喃(PBDFs)是常用溴代阻燃剂多溴联苯醚(PBDEs)转化过程的特征副产物,具有更强的毒性,可以在PBDEs的降解、含PBDEs的废弃物的处理以及一般工业过程中产生。目前的研究主要关注废弃物处理和工业热过程形成的PBDFs污染,然而PBDEs化学降解过程也可能是PBDFs的重要污染源。光解、零价铁还原、光催化和氧化都是PBDEs降解的有效方法,其中,光解会显著生成PBDFs。过去对PBDEs光解的研究主要关注其降解效率和生成低溴代PBDEs的逐步脱溴机制,而对副产物PBDFs探讨较少。目前普遍认为C-Br键的断裂是PBDEs光解的第一步,生成的PBDEs自由基夺取氢供体(如溶剂分子)的氢原子生成低溴代PBDEs,或发生分子内环化生成PBDFs。PBDFs已经在不同溶液中的PBDEs光解过程中普遍检出,生成量和溶液类型相关,然而,影响低溴代PBDEs和PBDFs生成比例的内在机理至今没有明确的解释。团队的前期工作已经揭示了邻位溴的存在是PBDEs光解生成PBDFs的前提条件,基于此,本研究结合动力学实验和理论计算,提出并验证了PBDEs光解过程中影响PBDFs生成比例的邻位碳自由基氢抽提和环化竞争反应机制,得到以下研究结论:
(1)以2,4,4'-三溴二苯醚(BDE-28)为模型污染物,通过密度泛函理论(DFT)计算获得了氢抽提和环化反应的速率常数,并利用竞争模型得到PBDFs的理论生成比例,成功解释了BDE-28在不同溶液中光解生成PBDFs的实验比例;
(2)DFT计算表明不同溶液中氢抽提反应速率常数差异显著,揭示了竞争过程中氢抽提反应的控制作用,即氢供体的供氢能力是PBDFs生成的关键控制因素,且与氢位点的结构特征相关。有机物饱和碳结构具有较强的供氢能力,不饱和碳结构以及含氧官能团的供氢能力较差,碳的级别越高或者连接了官能团其供氢能力越强,氢键的形成则弱化了分子的供氢能力。实验表明BDE-28在四氢呋喃、醇、饱和烃等溶剂中的PBDFs生成量较低,水的加入显著增加了有机溶液中PBDFs的生成,与计算的结果一致;
(3)在纯水体系中,由于水分子极差的供氢能力,BDE-28大部分转化为PBDFs。研究进一步考察了简单溶解性有机物(乙酸、柠檬酸、葡萄糖和丙氨酸)和大分子溶解性有机质(腐殖酸和富里酸)对PBDFs生成的影响。虽然DFT计算结果表明简单溶解性有机物表现出显著的供氢能力,但是低浓度条件下(<1 mmol/L)其在与环化反应的竞争中处于显著弱势,BDE-28仍然大部分转化为PBDFs,这与实验的结果一致。对于腐殖酸和富里酸,其对BDE-28的分配作用使BDE-28处于有机质大分子内的有机微环境中,内部表现出与有机溶剂相似的供氢能力,氢抽提反应优于环化反应,构建的分配-转化耦合方程较好地描述了不同腐殖酸/富里酸浓度下BDE-28的环化比率,腐殖酸/富里酸的供氢能力及其对BDE-28的分配作用共同决定了PBDFs的生成;
(4)从209种PBDEs中筛选出189种具有邻位溴的结构,确定了其在纯水中的512条环化和319条氢抽提反应路径,结合DFT计算和定量构效模型获得反应速率常数,利用竞争模型评估了189种PBDEs生成PBDFs的潜力,发现在无有机氢供体存在下,几乎所有脱邻位溴生成的自由基都大部分转化为PBDFs,表明在水环境中,一旦PBDEs邻位碳自由基生成,就有大部分转化为PBDFs的风险。
研究成果将为PBDEs的环境治理、转化和风险评估提供参考。
【入选期刊封面】

论文以“Hydrogen-Donor-Controlled Polybrominated Dibenzofuran (PBDF) Formation from Polybrominated Diphenyl Ether (PBDE) Photolysis in Solutions: Competition Mechanisms of Radical-Based Cyclization and Hydrogen Abstraction Reactions”为题发表于环境领域Top期刊Environmental Science & Technology(https://doi.org/10.1021/acs.est.2c08003),博士生杜晓冻同学主力贡献。欢迎感兴趣的同行关注。
