Platelets play an essential role in thrombosis and hemostasis. Abnormal hemostasis can cause spontaneous or severe post-traumatic bleeding. Bernard-Soulier syndrome (BSS) is a rare inherited bleeding disorder caused by a complete quantitative deficiency in the GPIb-IX-V complex(1-3). Multiple mutations in GP9 lead to the clinical manifestation of BSS(4-6). Understanding the roles and underlying mechanisms of GP9 in thrombopoiesis and establishing a proper animal model of BSS would be valuable to understand the disease pathogenesis and to improve its medical management.
On August 19, 2021, Yiyue Zhang's research group, both from South China University of Technology, publishes a research paper titled Establishment of a Bernard-Soulier syndrome model in zebrafish on Haematologica. This study established an inherited BSS zebrafish model and verified the clinical GP9 mutations by in vivo functional assay and tested clinical drugs for increasing platelets. The inherited BSS zebrafish model could be of benefit for in vivo verification of patient derived GP9 variants of uncertain significance and for potential BSS therapeutic strategy development.

To explicit the role of gp9 in thrombocytopoiesis, the research group generated a gp9-deficient zebrafish mutant by targeting exon 2 of the gp9 gene using the CRISPR-Cas9 technology (Figure 1A). They obtained an F0 mutant line containing gp9 (-17 bp, +0) (Figure 1B and C), and outcrossed this line with wild type zebrafish to generate F1 heterozygous and F2 homozygous progenies. The gp9 (-17 bp, +0) mutant was predicted to encode a truncated Gp9 protein lacking the two-leucine rich repeat domains and the transmembrane domain (Figure 1D). By detecting the gp9 mRNA by quantitative real-time polymerase chain reaction (qPCR), we found that the mutant fish produced only the mutated mRNA (Figure 1E).

Figure 1. Generation and characterization of gp9SMU15 zebrafish using CRISPR/Cas9.
To characterize the effects of gp9 disruption on thrombocytopoiesis in zebrafish, the research group detected thrombocyte numbers in gp9SMU15 mutants and their siblings by monitoring cd41:eGFP expression in Tg(cd41:eGFP) line, as the vast majority of circulating thrombocytes could be recognized by flow cytometry analysis (FACS) with brightly cd41:GFPhigh fluorescence in adult fish. Notably, although gp9SMU15 mutants were viable and able to survive to adulthood, the gp9SMU15 adult mutants displayed thrombocytopenia as the number of cd41:eGFPhigh(7) circulating thrombocytes in adult mutants was about 78% of those in adult WT sibling fish (Figure 2A and B). Next, they found a significant expansion of the precursor cell population in adult gp9SMU15 fish (Figure 2C-F). The above data demonstrated that gp9SMU15 zebrafish mutants probably had a developmental block, with expansion of larger thrombocytes and reduced small-nucleated thrombocytes in adulthood. BSS patients display increased bone marrow megakaryocytes but reduced mature platelets. In their study, zebrafish gp9SMU15 mutants showed a similar expansion of progenitor population but reduced circulating thrombocytes.
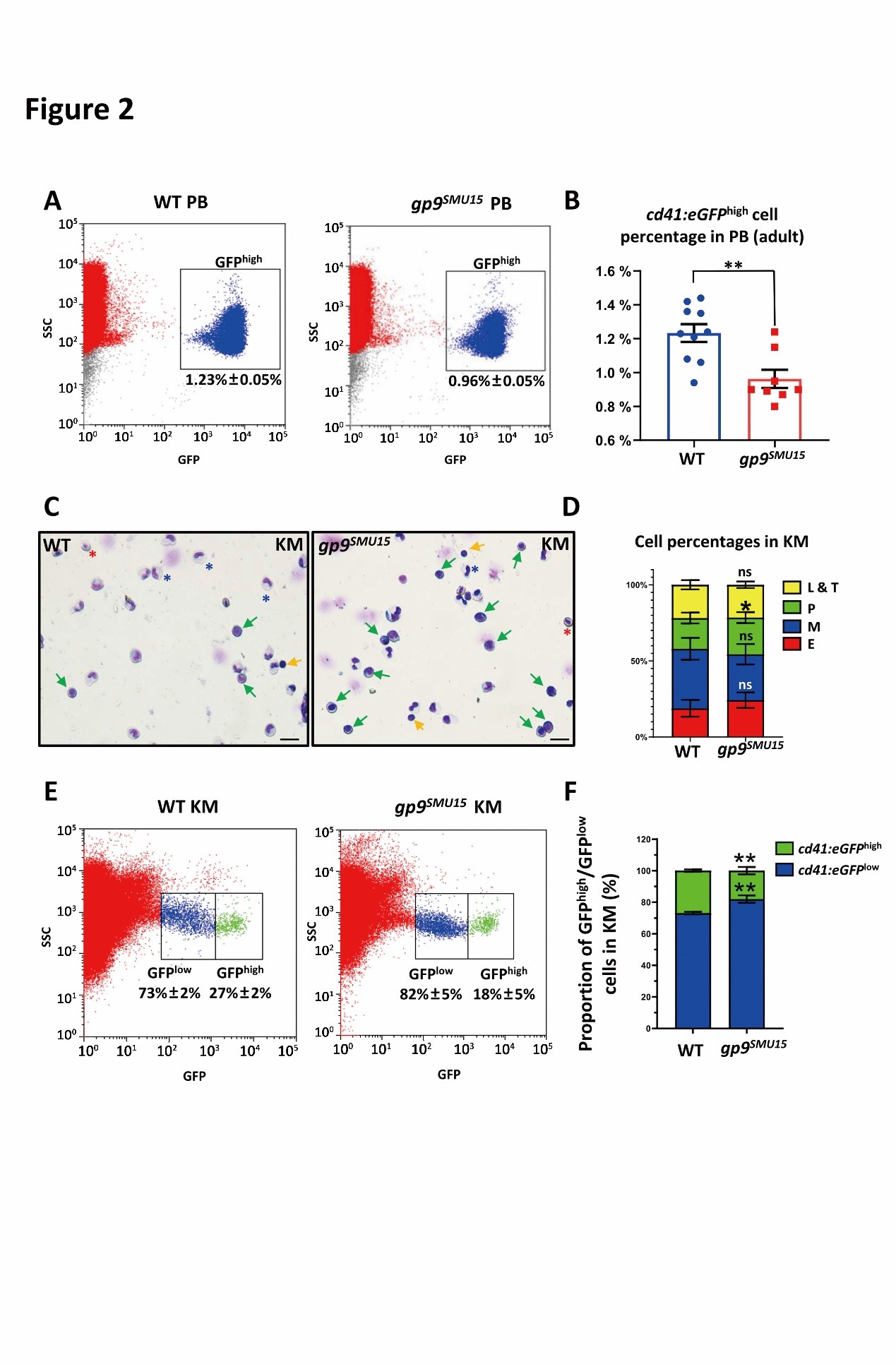
Figure 2. gp9SMU15 adult fish display thrombocytopenia and abnormal precursors expansion.
BSS patients generally have moderate thrombocytopenia, giant platelets and a bleeding tendency. They may suffer severe hemorrhage following injury or during surgery. Under normal condition, WT zebrafish rarely hemorrhage, whereas ~21% (4/19) gp9SMU15 mutants had blood spots on the body or near the tail fins (Figure 3A), indicating that gp9 mutation led to a high bleeding tendency. By calculating the time to stop bleeding after cut-tail injury, the research group found that the average time in gp9SMU15 adult mutants was ~50 seconds, much longer than in the wild type group (~20 seconds) (Figure 3B and C). They further used NaOH for chemical damage to induce gills bleeding and found a more severe gill bleeding in gp9SMU15 fish than in WT fish (Figure 3D and E). They then monitored the accumulation of cd41:eGFPhigh thrombocytes using FeCl3 to induce a vascular endothelium oxidative injury, and found that the time to accumulation of thrombocytes at the injured venule in gp9SMU15 zebrafish larva was significantly longer than in the WT sibling controls (Figure 3F). Taken together, these results demonstrated that the thrombocytopenic gp9SMU15 zebrafish exhibited bleeding disorders, similar to the extended clinical manifestations of clotting and prolonged hemorrhagic time in BSS patients.
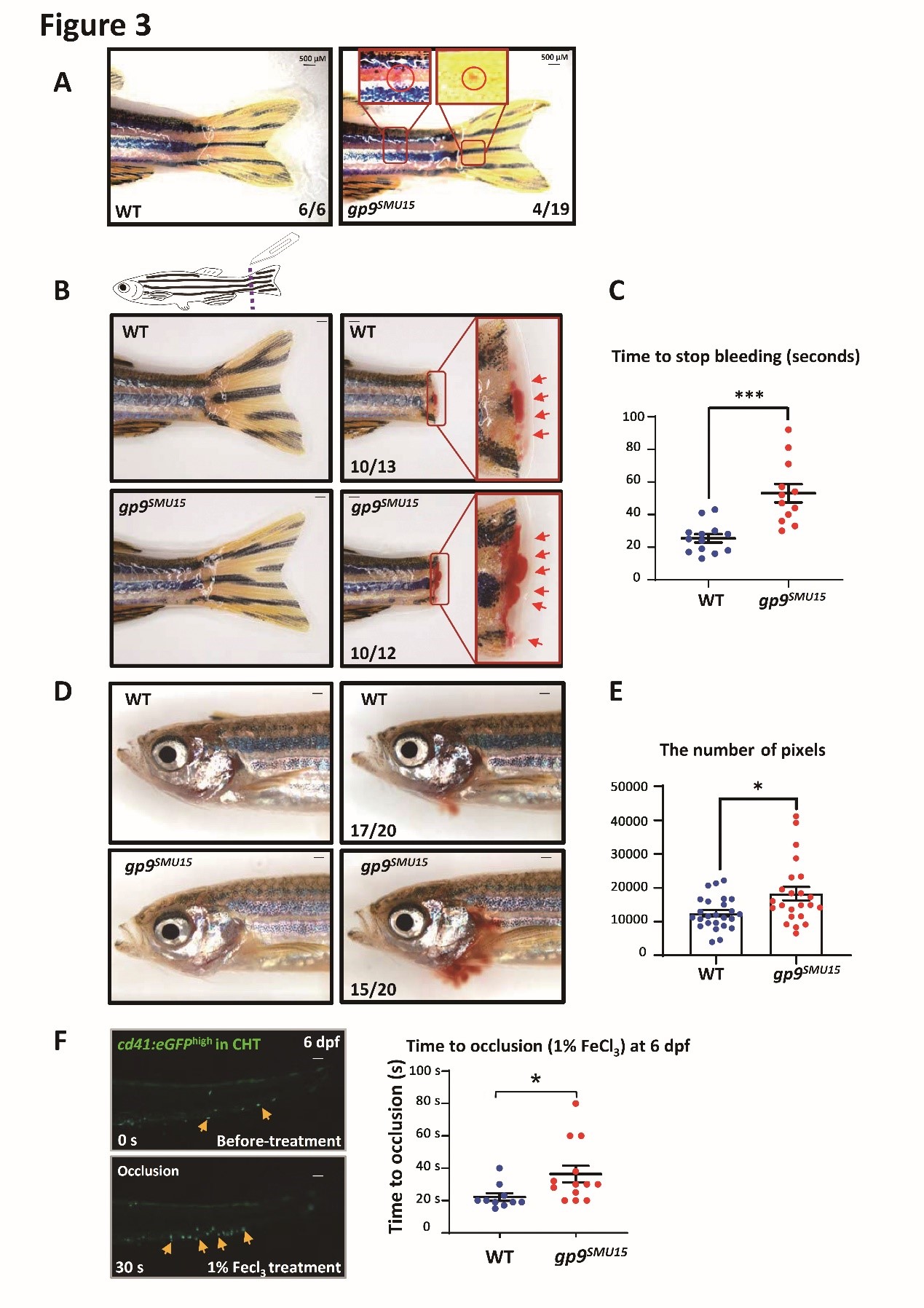
Figure 3. gp9SMU15 mutants display impaired thrombocyte function.
To characterize the effects of gp9 disruption on thrombocytopoiesis in zebrafish embryos or larvae, the authors detected thrombocyte numbers in gp9SMU15 mutants and their siblings by monitoring cd41 mRNA and cd41:eGFP expression (6-8). They isolated GFP+ cells from 3-dpf embryos by FACS, and found that the cd41:eGFPhigh cell population was significantly decreased but the cd41:eGFPlow cell population was increased in 3-dpf gp9SMU15 embryos (Figure 4A and B), suggesting that thrombocyte differentiation was affected by gp9 mutation. Furthermore, the quantification analysis showed that signals of cd41:eGFPhigh cells were markedly reduced in the caudal hematopoietic tissue (CHT) region of gp9SMU15 mutant larvae at 4 dpf compared with WT sibling embryos (Figure 4C and D), and several thrombocyte related genes, such as cd41 and genes required for thrombocytopoiesis (fog1 and nfe2) (Figure 4E). Notably, the aorta-gonad-mesonephros (AGM) cd41:eGFPlow cells, mainly represented for HSPCs, were unaltered in 2-dpf gp9SMU15 mutants (Figure 4F), and cmyb expression was unaltered in the AGM and CHT regions (Figure 4G), suggesting that gp9SMU15 mutants exhibited a lineage specific thrombocytic deficiency without affecting other blood lineage cells.
To explain the cellular basis of the thrombocytopenia in gp9SMU15 mutants, the research group further explored whether thrombocyte proliferation is reduced or apoptosis increased in gp9-deficient mutants. The results of BrdU and TUNEL indicated that thrombocytopenia could be mainly attributed to reduced proliferation but not likely caused by apoptosis.
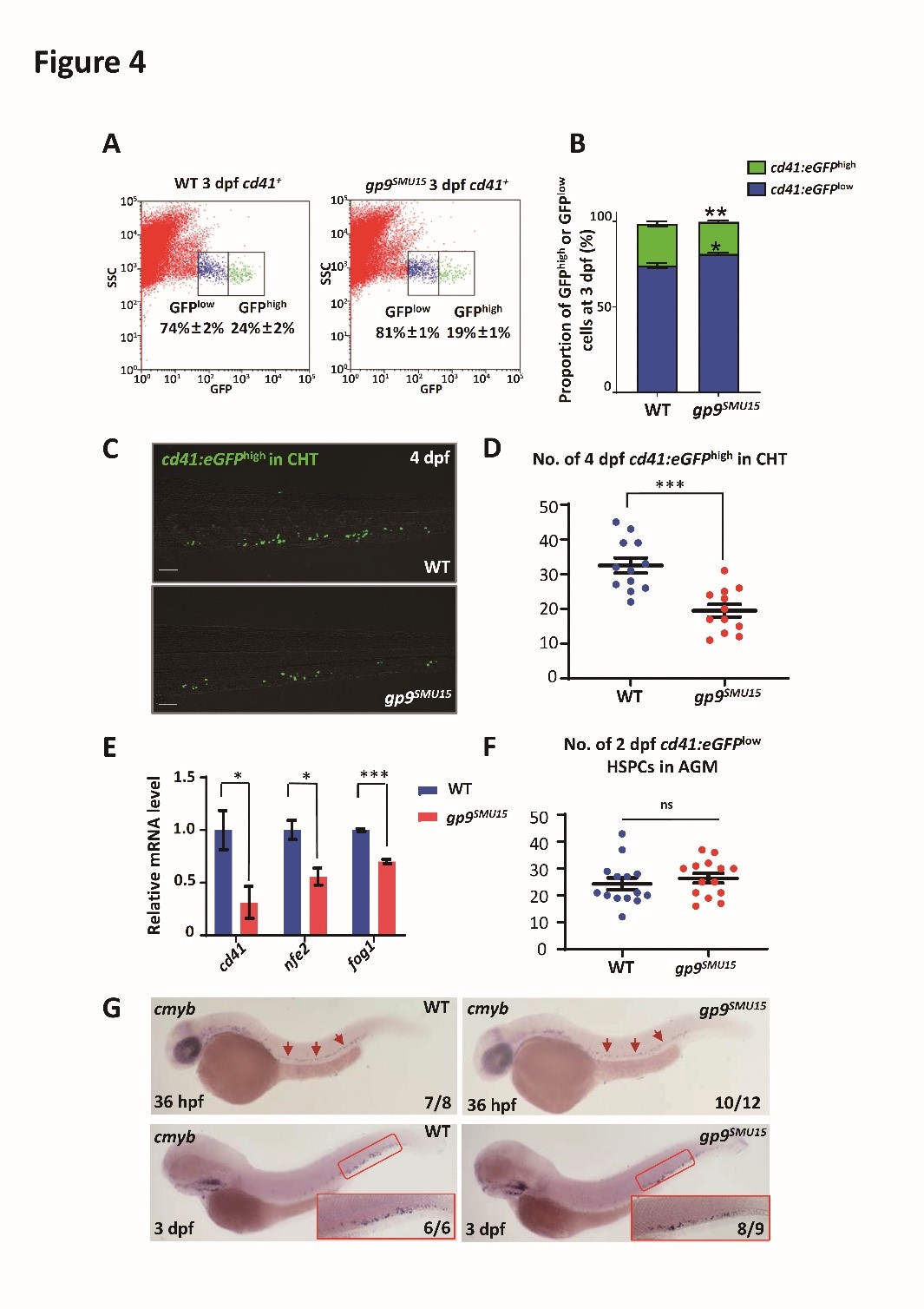
Figure 4. Thrombocytopenia in gp9SMU15 zebrafish embryo.
The authors further confirmed the functional conservation of thrombocytopoiesis in zebrafish and human GP9, and suggested that gp9SMU15 zebrafish could be utilized as a BSS model for fast-validating the functional significance of hsGP9 variants. We further overexpressed the human BSS isoform GP9c.70T>C and c.182A>G mRNA in gp9SMU15 zebrafish embryos to evaluate the function of the two isoforms on thrombocytopoiesis. Unlike hsGP9WT overexpression, hsGP970T>C and hsGP9182A>G overexpression could not restore cd41:eGFPhigh thrombocyte counts in gp9SMU15 mutants (Figure 5A), suggesting that two human GP9 mutations affect GP9 function on thrombocytopoiesis. Therefore, they showed that the rhTPO and decitabine could efficiently relapse BSS phenotypes in zebrafish larvae. The zebrafish BSS model, as an example for proving the principle, together with other established thrombocyte related zebrafish models, will allow us to examine the effect of clinical discovered mutations with uncertain clinical significance in thrombocyte development and function, as well as to establish a robust drug evaluation or screening platform to evaluate the therapeutic response of BSS, particularly against mutations with thrombocytopenia.
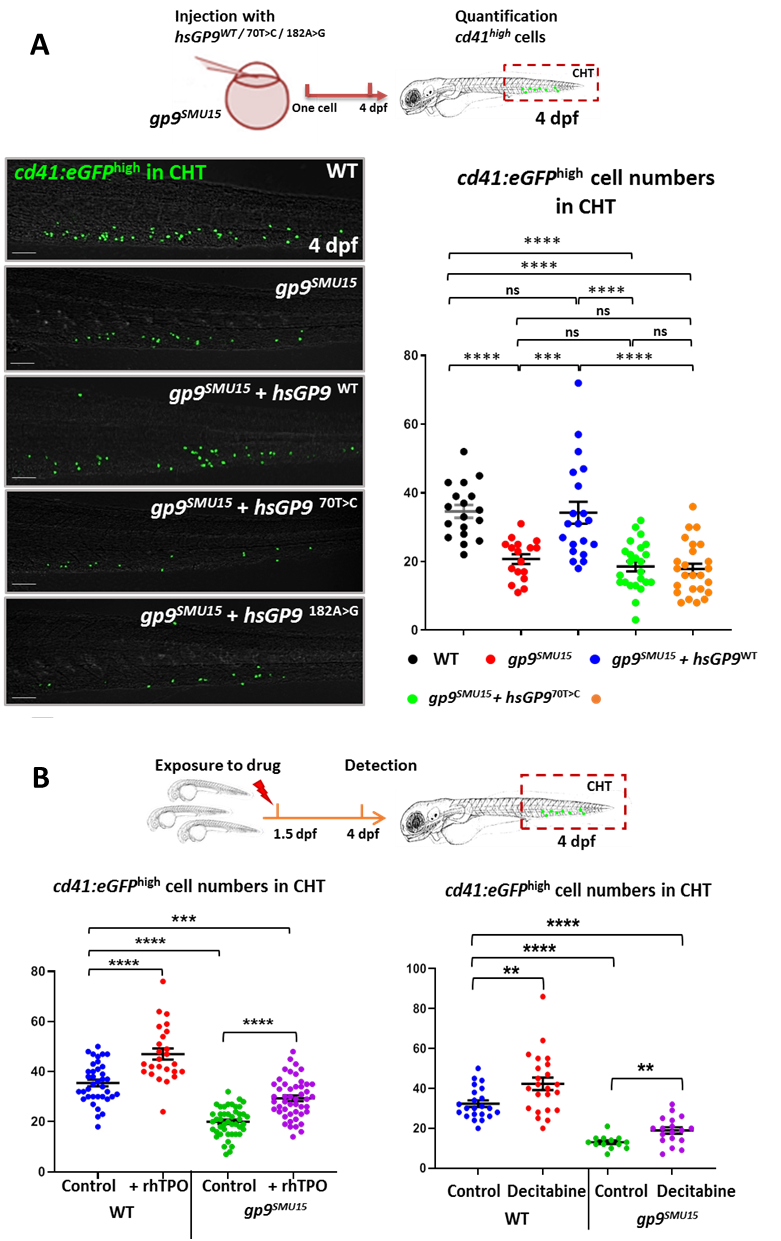
Figure 5. Application of BSS zebrafish disease model
Dr. Qing Lin, from South China University of Technology, Riyang Zhou, a doctoral student from Southern Medical University and Panpan Meng a doctoral student, from South China University of Technology are the co-first authors of the paper. Professor Yiyue Zhang from South China University of Technology is the corresponding author of the paper. This work was supported by National Natural Science Foundation of China (81870100 and 31871475), the National Key R&D Program of China (2018YFA0800200), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2019), and the Fundamental Research Funds for the Central Universities (2019ZD54 and 2018MS69).
Reference
1.Bernard J SJ: Sur une nouvelle variété de dystrophie thrombocytaire-hémorragipare congénitale. Sem Hop Paris 1948, 24(1948):3217-3222.
2.de la Salle C, Lanza F, Cazenave JP: Biochemical and molecular basis of Bernard-Soulier syndrome: a review. Nouvelle revue francaise d'hematologie 1995, 37(4):215.
3.Lopez J, Andrews R, Afshar-Kharghan V, Berndt M: Bernard-Soulier Syndrome. Blood 1998, 91:4397-4418.
4.Lanza F: Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet J Rare Dis 2006, 1(1):46-46.
5.Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, Caliskan U, Tokgoz H, Pecci A, Noris P et al: Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat 2014, 35(9):1033-1045.
6.McEwan PA, Yang W, Carr KH, Mo X, Zheng X, Li R, Emsley J: Quaternary organization of GPIb-IX complex and insights into Bernard-Soulier syndrome revealed by the structures of GPIbβ and a GPIbβ/GPIX chimera. Blood 2011, 118(19):5292-5301.
7.Lin H-F, Traver D, Zhu H, Dooley K, Paw BH, Zon LI, Handin RI: Analysis of thrombocyte development in CD41-GFP transgenic zebrafish. Blood 2005, 106(12):3803-3810.
8.Ma D, Zhang J, Lin HF, Italiano J, Handin RI: The identification and characterization of zebrafish hematopoietic stem cells. Blood 2011, 118(2):289-297.
